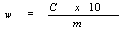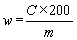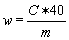S.I. No. 269/1994 - European Communities (Feeding Stuffs) (Methods of Analysis) (Amendment) Regulations, 1994.
S.I. No. 269 of 1994. | |||||||||||||||||||||||||||||||||||||||||||||||
EUROPEAN COMMUNITIES (FEEDING STUFFS) (METHODS OF ANALYSIS) (AMENDMENT) REGULATIONS, 1994. | |||||||||||||||||||||||||||||||||||||||||||||||
I, JOE WALSH, Minister for Agriculture, Food and Forestry, in exercise of the powers conferred on me by section 3 of the European Communities Act, 1972 (No. 27 of 1972), and for the purposes of giving effect to the Eleventh Commission Directive No. 93/70/EEC of 28th July, 19931 and the Twelfth Commission Directive No. 93/117/EC of 17th December, 19932, hereby make the following Regulations. | |||||||||||||||||||||||||||||||||||||||||||||||
1 O.J. No. L 234, 17.9.1993, P. 17. | |||||||||||||||||||||||||||||||||||||||||||||||
2 O.J. L 329, 30.12.1993, P. 54. 1. (1) These Regulations may be cited as the European Communities (Feedingstuffs) (Methods of Analysis) (Amendment) Regulations, 1994. | |||||||||||||||||||||||||||||||||||||||||||||||
(2) The European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1978 to 1993, and these Regulations may be cited together as the European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1978 to 1994. 2. In these Regulations: | |||||||||||||||||||||||||||||||||||||||||||||||
"the Principal Regulations" means the European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1978 ( S.I. No. 250 of 1978 ); | |||||||||||||||||||||||||||||||||||||||||||||||
"the Regulations of 1985" means the European Communities (Feeding Stuffs) (Methods of Analysis) (Amendment) Regulations, 1985 ( S.I. No. 16 of 1985 ). 3. The Principle Regulations are hereby amended by:— | |||||||||||||||||||||||||||||||||||||||||||||||
( a ) the substitution of the following Regulation for Regulation 2: | |||||||||||||||||||||||||||||||||||||||||||||||
"2. The methods of analysis specified in Part II of these Regulations shall be the methods of analysis to be used for the purposes of the European Communities (Feeding Stuffs) (Additives) Regulations, 1989 ( S.I. No. 49 of 1989 ) and subsequent amendments thereto, the European Communities (Feedingstuffs) (Tolerances of Undesirable Substances and Products) Regulations, 1989 ( S.I. No. 216 of 1989 ) and subsequent amendments thereto, and the European Communities (Protein Feedingstuffs) Regulations, 1986 ( S.I. No. 433 of 1986 ), and subsequent amendments thereto."; | |||||||||||||||||||||||||||||||||||||||||||||||
( b ) the insertion of the following paragraphs in Part II after paragraph 30 (inserted by Regulation 2 (c) of the Regulations of 1985: | |||||||||||||||||||||||||||||||||||||||||||||||
"31Determination of halofuginone | |||||||||||||||||||||||||||||||||||||||||||||||
DL-trans-7-bromo-6-chloro-3-[3-(3-hydroxy-2-piperidyl)acetonyl]-quinazolin-4-(3H)—one hydrobromide. | |||||||||||||||||||||||||||||||||||||||||||||||
1. Purpose and scope | |||||||||||||||||||||||||||||||||||||||||||||||
To determine the content of halofuginone in feedingstuffs. The lower limit of determination is 1 mg/kg. | |||||||||||||||||||||||||||||||||||||||||||||||
2. Principle | |||||||||||||||||||||||||||||||||||||||||||||||
After treatment with hot water, halofuginone is extracted as the free base into ethyl acetate and subsequently partitioned as the hydrochloride into an aqueous acid solution. The extract is purified by ion-exchange chromatography. The content of halofuginone is determined by reserved-phase high performance liquid chromatography (HPLC) using a UV detector. | |||||||||||||||||||||||||||||||||||||||||||||||
3. Reagents | |||||||||||||||||||||||||||||||||||||||||||||||
3.1 Acetonitrile, HPLC grade. | |||||||||||||||||||||||||||||||||||||||||||||||
3.2 Amberlite XAD-2 resin. | |||||||||||||||||||||||||||||||||||||||||||||||
3.3 Ammonium acetate. | |||||||||||||||||||||||||||||||||||||||||||||||
3.4 Ethyl acetate. | |||||||||||||||||||||||||||||||||||||||||||||||
3.5 Acetic acid, glacial. | |||||||||||||||||||||||||||||||||||||||||||||||
3.6 Halofuginone standard substance (DL-trans-7—bromo-6-chloro-3-[3-hydroxy-2-piperidyl)acetonyl]-quinazolin-4-(3H)-one hydrobromide, E 764). | |||||||||||||||||||||||||||||||||||||||||||||||
3.6.1 Halofuginone stock standard solution 100 µg/ml. | |||||||||||||||||||||||||||||||||||||||||||||||
Weight to the nearest 0.1 mg, 50 mg of halofuginone (3.6) in a 500 ml graduated flask, dissolve in ammonium acetate buffer solution (3.18), make up to the mark with the buffer solution and mix. This solution is stable for three weeks at 5°C if stored in the dark. | |||||||||||||||||||||||||||||||||||||||||||||||
3.6.2 Calibration solutions. | |||||||||||||||||||||||||||||||||||||||||||||||
Into a series of 100 ml graduated flasks transfer 1.0, 2.0, 3.0, 4.0 and 6.0 ml of the stock standard solution (3.6.1). Make up to the mark with mobile phase (3.21) and mix. Thsse solutions have concentrations of 1.0, 2.0, 3.0, 4.0 and 6.0 µg/ml of halofuginone respectively. These solutions must be freshly prepared before use. | |||||||||||||||||||||||||||||||||||||||||||||||
3.7 Hydrochloric acid (p20 approximately 1.16 g/ml). | |||||||||||||||||||||||||||||||||||||||||||||||
3.8 Methanol. | |||||||||||||||||||||||||||||||||||||||||||||||
3.9 Silver nitrate. | |||||||||||||||||||||||||||||||||||||||||||||||
3.10 Sodium ascorbate. | |||||||||||||||||||||||||||||||||||||||||||||||
3.11 Sodium carbonate. | |||||||||||||||||||||||||||||||||||||||||||||||
3.12 Sodium chloride. | |||||||||||||||||||||||||||||||||||||||||||||||
3.13 EDTA (ethylenediaminetetraacetic acid, disodium salt). | |||||||||||||||||||||||||||||||||||||||||||||||
3.14 Water, HPLC grade. | |||||||||||||||||||||||||||||||||||||||||||||||
3.15 Sodium carbonate solution, ß = 10 g/100 ml. | |||||||||||||||||||||||||||||||||||||||||||||||
3.16 Sodium chloride-saturated sodium carbonate solution, ß = 5 g/100 ml. | |||||||||||||||||||||||||||||||||||||||||||||||
Dissolve 50 g of sodium carbonate (3.11) in water, dilute to 1 l and add sodium chloride (3.12) until the solution is saturated. | |||||||||||||||||||||||||||||||||||||||||||||||
3.17 Hydrochloric acid, approximately 0.1 mol/l. | |||||||||||||||||||||||||||||||||||||||||||||||
Dilute 10 ml of HC1 (3.7) with water to 1 l. | |||||||||||||||||||||||||||||||||||||||||||||||
3.18 Ammonium acetate buffer solution, approximately 0.25 mol/l. | |||||||||||||||||||||||||||||||||||||||||||||||
Dissolve 19.3 g of ammonium acetate (3.3) and 30 ml of acetic acid (3.5) in water (3.14) and dilute to 1 l. | |||||||||||||||||||||||||||||||||||||||||||||||
3.19 Amberlite XAD-2 resin preparation. | |||||||||||||||||||||||||||||||||||||||||||||||
Wash an appropriate quantity of Amberlite (3.2) with water until all chloride ions have been removed, as indicated by a silver nitrate (3.20) test performed on the discarded aqueous phase. Then wash the resin with 50 ml of methanol (3.8), discard the methanol and store the resin under fresh methanol. | |||||||||||||||||||||||||||||||||||||||||||||||
3.20 Silver nitrate solution, approximately 0.1 mol/l. | |||||||||||||||||||||||||||||||||||||||||||||||
Dissolve 0.17 g of silver nitrate (3.9) in 10 ml of water. | |||||||||||||||||||||||||||||||||||||||||||||||
3.21 HPLC Mobile phase. | |||||||||||||||||||||||||||||||||||||||||||||||
Mix 500 ml of acetonitrile (3.1) with 300 ml of ammonium acetate buffer solution (3.18) and 1 200 ml of water (3.14). Adjust the pH to 4.3 using acetic acid (3.5). Filter through a 0.22 µm filter (4.8) and degas the solution (e.g. by ultrasonification for 10 minutes). This solution is stable for one month, if stored in the dark in a closed container. | |||||||||||||||||||||||||||||||||||||||||||||||
4. Apparatus | |||||||||||||||||||||||||||||||||||||||||||||||
4.1 Ultrasonic bath. | |||||||||||||||||||||||||||||||||||||||||||||||
4.2 Rotary film evaporator. | |||||||||||||||||||||||||||||||||||||||||||||||
4.3 Centrifuge. | |||||||||||||||||||||||||||||||||||||||||||||||
4.4 HPLC equipment with variable wavelength ultraviolet detector or diode-array detector. | |||||||||||||||||||||||||||||||||||||||||||||||
4.4.1 Liquid chromatographic column, 300 mm x 4 mm, C 18, 10 µm packaging, or an equivalent column. | |||||||||||||||||||||||||||||||||||||||||||||||
4.5 Glass column (300 mm x 10 mm) fitted with a sintered-glass filter and a stopcock. | |||||||||||||||||||||||||||||||||||||||||||||||
4.6 Glass-fibre filters, diameter 150 mm. | |||||||||||||||||||||||||||||||||||||||||||||||
4.7 Membrane filters, 0.45 µ.m. | |||||||||||||||||||||||||||||||||||||||||||||||
4.8 Membrane filters, 0.22 µ.m. | |||||||||||||||||||||||||||||||||||||||||||||||
5. Procedure | |||||||||||||||||||||||||||||||||||||||||||||||
Note: Halofuginone as the free base is unstable in alkaline and ethyl acetate solutions. It should not remain in ethyl acetate for more than 30 minutes. | |||||||||||||||||||||||||||||||||||||||||||||||
5.1 General. | |||||||||||||||||||||||||||||||||||||||||||||||
5.1.1 A blank feed should be analysed to check that neither halofuginone nor interfering substances are present. | |||||||||||||||||||||||||||||||||||||||||||||||
5.1.2 A recovery test should be carried out by analysing the blank feed which has been fortified by addition of a quantity of halofuginone, similar to that present in the sample. To fortify at a level of 3 mg/kg, add 300 µl of the stock standard solution (3.6.1) to 10 g of the blank feed, mix and wait for 10 minutes before proceeding with the extraction step (5.2). | |||||||||||||||||||||||||||||||||||||||||||||||
Note: for the purpose of this method, the blank feed should be similar in type to that of the sample and on analysis halofuginone should not be detected. | |||||||||||||||||||||||||||||||||||||||||||||||
5.2 Extraction. | |||||||||||||||||||||||||||||||||||||||||||||||
Weigh to the nearest 0.1 g, 10 g of the prepared sample, into a 200 ml centrifuge tube, add 0,5 g of sodium ascorbate (3.10), 0.5 g of EDTA (3.13) and 20 ml of water and mix. Place the tube for 5 minutes in a water bath (80°C). After cooling down to room temperature, add 20 ml of sodium carbonate solution (3.15) and mix. Add immediately 100 ml of ethyl acetate (3.4) and shake vigorously by hand for 15 seconds. Then place the tube for three minutes in the ultrasonic bath (4.1) and loosen the stopper. Centrifuge for two minutes and decant the ethyl acetate phase through a glass fibre filter (4.6), into a 500 ml separating funnel. Repeat the extraction of the sample with a second portion of 100 ml of ethyl acetate. Wash the combined extracts for one minute with 50 ml of sodium chloride-saturated sodium carbonate solution (3.16) and discard the aqueous layer. | |||||||||||||||||||||||||||||||||||||||||||||||
Extract the organic layer for 1 minute with 50 ml of hydrochloric acid (3.17). Run the lower acid layer into a 250 ml separating funnel. Re-extract the organic layer for 1.5 minutes with a further 50 ml of hydrochloric acid and combine with the first extract. Wash the combined acid extracts by swirling for approximately 10 seconds with 10 ml of ethyl acetate (3.4). Quantitatively transfer the aqueous layer into a 250 ml round-bottomed flask and discard the organic phase. Evaporate all the remaining ethyl acetate from the acid solution using a rotary film evaporator (2.4). The temperature of the water bath should not exceed 40°C. Under a vacuum of approximately 25 mbar all of the residual ethyl acetate will be removed within 5 minutes at 38°C. | |||||||||||||||||||||||||||||||||||||||||||||||
5.3 Clean up. | |||||||||||||||||||||||||||||||||||||||||||||||
5.3.1 Preparation of the Amberlite column. | |||||||||||||||||||||||||||||||||||||||||||||||
An XAD-2 column is prepared for each sample extract. Transfer 10 g of prepared Amberlite (3.19) into a glass column (4.5) with methanol (3.8). Add a small plug of glass-wool to the top of the resin bed. Drain the methanol from the column and wash the resin with 100 ml of water, stopping the flow as the liquid reaches the top of the resin bed. Allow the column to equilibrate for 10 minutes before use. Never allow the column to run dry. | |||||||||||||||||||||||||||||||||||||||||||||||
5.3.2 Sample clean up. | |||||||||||||||||||||||||||||||||||||||||||||||
Transfer the extract (5.2) quantitatively to the top of the prepared Amberlite column (5.3.1) and elute, discarding the eluate. The rate of elution should not exceed 20 ml/min. Rinse the round-bottomed flask with 20 ml of hydrochloric acid (3.17) and use this to wash the resin column. Blow through any remaining acid solution with a stream of air. Discard the washings. Add 100 ml of methanol (3.8) to the column and allow 5 to 10 ml to elute, collecting the eluate in a 250 ml round-bottomed flask. Leave the remaining methanol for 10 minutes to equilibrate with the resin and continue the elution at a rate not exceeding 20 ml/min, collecting the eluate in the same round-bottomed flask. Evaporate the methanol on the rotary film evaporator (4.2), the temperature of the waterbath should not exceed 40°C. Transfer the residue quantitatively into a 10 ml calibrated flask using the mobile phase (3.21). Make up to the mark with mobile phase and mix. An aliquot is filtered through a membrane filter (4.7). Reserve this solution for the HPLC determination (5.4). | |||||||||||||||||||||||||||||||||||||||||||||||
5.4 HPLC determination. | |||||||||||||||||||||||||||||||||||||||||||||||
5.4.1 Parameters. | |||||||||||||||||||||||||||||||||||||||||||||||
The following conditions are offered for guidance, other conditions may be used provided they yield equivalent results. | |||||||||||||||||||||||||||||||||||||||||||||||
Liquid chromatographic column (4.1.1). | |||||||||||||||||||||||||||||||||||||||||||||||
HPLC Mobile phase (3.21). | |||||||||||||||||||||||||||||||||||||||||||||||
Flow rate: 1.5 to 2 ml/min. | |||||||||||||||||||||||||||||||||||||||||||||||
Detection wavelength: 243 nm. | |||||||||||||||||||||||||||||||||||||||||||||||
Injection volume: 40 to 100 µl. | |||||||||||||||||||||||||||||||||||||||||||||||
Check the stability of the chromatographic system, by injecting the calibration solution prepared in (3.6.2) and which contains 3.0 µg/ml, several times until constant peak heights (or areas) and retention times are achieved. | |||||||||||||||||||||||||||||||||||||||||||||||
5.4.2 Calibration graph. | |||||||||||||||||||||||||||||||||||||||||||||||
Inject each of the calibration solutions prepared in (3.6.2) several times and measure the peak heights (or areas) for each concentration. Plot a calibration graph using the mean peak heights or areas of the calibration solutions as the ordinates and the corresponding concentrations in µg/ml as the abscissae. | |||||||||||||||||||||||||||||||||||||||||||||||
5.4.3 Sample solution. | |||||||||||||||||||||||||||||||||||||||||||||||
Inject the sample extract (5.3.2) several times, using the same volume as taken for the calibration solutions and determine the mean peak heights (or area) of the halofuginone peaks. | |||||||||||||||||||||||||||||||||||||||||||||||
6. Calculation of results | |||||||||||||||||||||||||||||||||||||||||||||||
Determine the concentration of the sample solutions in µg/ml, from the mean height (area) of the halofuginone peaks of the sample solution by reference to the calibration graph (5.4.2). | |||||||||||||||||||||||||||||||||||||||||||||||
The content of halofuginone w (mg/kg) of the sample is given by the following formula: | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
in which: | |||||||||||||||||||||||||||||||||||||||||||||||
— c: halofuginone concentration of the sample solution in µg/ml. | |||||||||||||||||||||||||||||||||||||||||||||||
— m: mass of the test portion in grams. | |||||||||||||||||||||||||||||||||||||||||||||||
7. Validation of the results | |||||||||||||||||||||||||||||||||||||||||||||||
7.1 Identity. | |||||||||||||||||||||||||||||||||||||||||||||||
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract and the calibration solution (3.6.2) which contains 6.0µg/ml are compared. | |||||||||||||||||||||||||||||||||||||||||||||||
7.1.1 Co-chromatography. | |||||||||||||||||||||||||||||||||||||||||||||||
A sample extract is fortified by addition of an appropriate amount of a calibration solution (3.6.2). The amount of added halofuginone should be similar to the estimated amount of halofuginone found in the sample extract. | |||||||||||||||||||||||||||||||||||||||||||||||
Only the height of the halofuginone peak should be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its maximum height, must be within ± 10% of the original width. | |||||||||||||||||||||||||||||||||||||||||||||||
7.1.2 Diode-array detection. | |||||||||||||||||||||||||||||||||||||||||||||||
The results are evaluated according to the following criteria: | |||||||||||||||||||||||||||||||||||||||||||||||
( a ) the wavelength of maximum absorption of the sample and of the standard spectra, recorded at the peak apex on the chromatogram, must be the same within a margin determined by the resolving power of the detection system. For diode-array detection, this is typically within ± 2 nm; | |||||||||||||||||||||||||||||||||||||||||||||||
( b ) between 225 and 300 nm, the sample and standard spectra recorded at the peak apex on the chromatogram, must not be different for those parts of the spectrum within the range 10 to 100% or relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15% of the absorbance of the standard analyte; | |||||||||||||||||||||||||||||||||||||||||||||||
( c ) between 225 and 300 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 to 100% of relative absorbance. This criterion is met when the same maxima are present and when at all observed points the deviation between the spectra does not exceed 15% of the absorbance of the spectrum of the apex. | |||||||||||||||||||||||||||||||||||||||||||||||
If one of these criteria is not met the presence of the analyte has not been confirmed. | |||||||||||||||||||||||||||||||||||||||||||||||
7.2 Repeatability. | |||||||||||||||||||||||||||||||||||||||||||||||
The difference between results of two parallel determinations carried out on the same sample must not exceed 0.5 mg/kg for halofuginone contents up to 3 mg/kg. | |||||||||||||||||||||||||||||||||||||||||||||||
7.3 Recovery. | |||||||||||||||||||||||||||||||||||||||||||||||
For the fortified blank sample the recovery should be at least 80%. | |||||||||||||||||||||||||||||||||||||||||||||||
8. Results of a collaborative study | |||||||||||||||||||||||||||||||||||||||||||||||
A collaborative study1 was arranged in which three samples were analysed by eight laboratories. The results are given in the Table below: | |||||||||||||||||||||||||||||||||||||||||||||||
1 The Analyst 108, 1983, pp. 1252 to 1256. | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
n.d.: not detected. | |||||||||||||||||||||||||||||||||||||||||||||||
SR: standard deviation of reproducibility. | |||||||||||||||||||||||||||||||||||||||||||||||
CVR: coefficient of variation. | |||||||||||||||||||||||||||||||||||||||||||||||
Rec: recovery. | |||||||||||||||||||||||||||||||||||||||||||||||
32. Determination of Robenidine. | |||||||||||||||||||||||||||||||||||||||||||||||
1,3—bis[(4-chlorobenzylidene)amino]guanidine-hydrochloride | |||||||||||||||||||||||||||||||||||||||||||||||
1. Purpose and scope | |||||||||||||||||||||||||||||||||||||||||||||||
To determine the content of robenidine in feedingstuffs. The lower limit of determination is 5 mg/kg. | |||||||||||||||||||||||||||||||||||||||||||||||
2. Principle | |||||||||||||||||||||||||||||||||||||||||||||||
The sample is extracted with acidified methanol. The extract is dried and an aliquot portion subjected to a clean-up on an aluminium oxide column. Robenidine is eluted from the column with methanol, concentrated, and made up to a suitable volume with mobile phase. The content of robenidine is determined by reversed-phase high-performance liquid chromatography (HPLC) using an UV detector. | |||||||||||||||||||||||||||||||||||||||||||||||
3. Reagents | |||||||||||||||||||||||||||||||||||||||||||||||
3.1 Methanol. | |||||||||||||||||||||||||||||||||||||||||||||||
3.2 Acidified methanol. | |||||||||||||||||||||||||||||||||||||||||||||||
Transfer 4.0 ml hydrochloric acid (p20 c. 1.18 g/ml) into a 500 ml graduated flask, make up to the mark with methanol (3.1) and mix. This solution should be freshly prepared before use. | |||||||||||||||||||||||||||||||||||||||||||||||
3.3 Acetonitrile, HPLC grade. | |||||||||||||||||||||||||||||||||||||||||||||||
3.4 Molecular sieve | |||||||||||||||||||||||||||||||||||||||||||||||
Type 3A, 8 to 12 mesh beads (1.6-2.5 mm beads, crystalline alumino-silicate, diameter of pores 0.3 mm) | |||||||||||||||||||||||||||||||||||||||||||||||
3.5 Aluminium oxide; acidic activity grade I for column chromatography. | |||||||||||||||||||||||||||||||||||||||||||||||
Transfer 100 g aluminium oxide into a suitable container and add 2.0 ml of water. Stopper and shake for approximately 20 minutes. Store in a well stoppered container. | |||||||||||||||||||||||||||||||||||||||||||||||
3.6 Potassium dihydrogen phosphate solution, c = 0.025 mol/l. | |||||||||||||||||||||||||||||||||||||||||||||||
Dissolve 3.40 g of potassium dihydrogen phosphate in water (HPLC grade) in a 1000 ml graduated flask, make up to the mark and mix. | |||||||||||||||||||||||||||||||||||||||||||||||
3.7 Di-sodium hydrogen phosphate solution, c = 0.025 mol/l. | |||||||||||||||||||||||||||||||||||||||||||||||
Dissolve 3.55 g of anhydrous (or 4.45 g of dehydrate or 8.95 g of dodecahydrate) di-sodium hydrogen phosphate in water (HPLC grade) in a 1 000 ml graduated flask, make up to the mark and mix. | |||||||||||||||||||||||||||||||||||||||||||||||
3.8 HLPC mobile phase. | |||||||||||||||||||||||||||||||||||||||||||||||
Mix together the following reagents: | |||||||||||||||||||||||||||||||||||||||||||||||
650 ml acetonitrile (3.3), | |||||||||||||||||||||||||||||||||||||||||||||||
250 ml water (HPLC-grade), | |||||||||||||||||||||||||||||||||||||||||||||||
50 ml potassium di-hydrogen phosphate solution (3.6), | |||||||||||||||||||||||||||||||||||||||||||||||
50 ml di-sodium hydrogen phosphate solution (3.7). | |||||||||||||||||||||||||||||||||||||||||||||||
Filter through a 0.22 µm filter (4.6) and degas the solution, (e.g. by ultrasonification for 10 minutes). | |||||||||||||||||||||||||||||||||||||||||||||||
3.9 Standard substance. | |||||||||||||||||||||||||||||||||||||||||||||||
Pure robenidine: 1,3—bis[4-chlorobenzylidene]amino] guanidine hydrochloride, E 750. | |||||||||||||||||||||||||||||||||||||||||||||||
3.9.1 Robenidine stock standard solution: 300 µg/ml: | |||||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest 0.1 mg, 30 mg of robenidine standard substance (3.9). Dissolve in acidified methanol (3.2) in a 100 ml graduated flask, make up to the mark with the same solvent and mix. Wrap the flask with aluminium foil and store in a dark place. | |||||||||||||||||||||||||||||||||||||||||||||||
3.9.2 Robenidine intermediate standard solution: 12 µg/ml. | |||||||||||||||||||||||||||||||||||||||||||||||
Transfer 10.0 ml of the stock standard solution (3.9.1) into a 250 ml graduated flash, make up to the mark with the mobile phase (3.8) and mix. Wrap the flask with aluminium foil and store in a dark place. | |||||||||||||||||||||||||||||||||||||||||||||||
3.9.3 Calibration solutions. | |||||||||||||||||||||||||||||||||||||||||||||||
Into a series of 50 ml calibrated flasks, transfer 5.0, 10.0, 15.0, 20.0 and 25.0 ml of the intermediate standard solution (3.9.2). Make up to the mark with mobile phase (3.8) and mix. These solutions correspond to 1.2, 2.4, 3.6, 4.8, and 6.0, µg/ml of robenidine respecitively. These solutions must be freshly prepared before use. | |||||||||||||||||||||||||||||||||||||||||||||||
4. Apparatus | |||||||||||||||||||||||||||||||||||||||||||||||
4.1 Glass column. | |||||||||||||||||||||||||||||||||||||||||||||||
Constructed of amber glass fitted with a stopcock and a reservoir of approximately 150 ml capacity, internal diameter 10 to 15 mm, length 250 mm. | |||||||||||||||||||||||||||||||||||||||||||||||
4.2 Laboratory wrist-action shaker. | |||||||||||||||||||||||||||||||||||||||||||||||
4.3 Rotary film evaporator. | |||||||||||||||||||||||||||||||||||||||||||||||
4.4 HPLC equipment with variable wavelength ultraviolet detector or diode array detector operating in the range of 250 to 400 mm. | |||||||||||||||||||||||||||||||||||||||||||||||
4.4.1 Liquid chromatographic column: 300 mm x 4 mm, C18, 10 µm packing or equivalent. | |||||||||||||||||||||||||||||||||||||||||||||||
4.5 Glass fibre filter paper (Whatman GF/A or equivalent). | |||||||||||||||||||||||||||||||||||||||||||||||
4.6 Membrane filters, 0.22 µm. | |||||||||||||||||||||||||||||||||||||||||||||||
4.7 Membrane filters, 0.45 µm. | |||||||||||||||||||||||||||||||||||||||||||||||
5. Procedure | |||||||||||||||||||||||||||||||||||||||||||||||
Note: Robenidine is light-sensitive. Amber glassware should be used in all operations. | |||||||||||||||||||||||||||||||||||||||||||||||
5.1 General. | |||||||||||||||||||||||||||||||||||||||||||||||
5.1.1 A blank feed should be analysed to check that neither robenidine nor interfering substances are present. | |||||||||||||||||||||||||||||||||||||||||||||||
5.1.2 A recovery test should be carried out by analysing the blank feed (5.1.1) which has been fortified by addition of a quantity of robenidine, similar to that present in the sample. To fortify at a level of 60 mg/kg, transfer 3.0 ml of the stock standard solution (3.9.1) to a 250 ml conical flask. Evaporate the solution to c. 0.5 ml in a stream of nitrogen. Add 15 g of the blank feed, mix and wait for 10 minutes before proceeding with the extraction step (5.2). | |||||||||||||||||||||||||||||||||||||||||||||||
Note: For the purpose of this method, the blank feed should be similar in type to that of the sample and on analysis robenidine should not be detected. | |||||||||||||||||||||||||||||||||||||||||||||||
5.2 Extraction. | |||||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest 0.01 g, approximately 15 g of the prepared sample. Transfer to a 250 ml conical flask and add 100.0 ml of acidified methanol (3.2), stopper and shake for one hour on the shaker (4.2). Filter the solution through a glass fibre filter paper (4.5) and collect the whole filtrate in a 150 ml conical flash. Add 7.5 g molecular sieve (3.4), stopper and shake for five minutes. Filter immediately through a glass-fibre filter paper. Retain this solution for the purification step(5.3). | |||||||||||||||||||||||||||||||||||||||||||||||
5.3 Purification. | |||||||||||||||||||||||||||||||||||||||||||||||
5.3.1 Preparation of the aluminium-oxide column. | |||||||||||||||||||||||||||||||||||||||||||||||
Insert a small glass-wool plug into the lower end of a glass column (4.1) and tamp it down using a glass rod. Weigh out 11.0 g of the prepared aluminium oxide (3.5) and transfer to the column. Care should be taken to minimise the exposure to the atmosphere during this stage. Gently tap the loaded column at its lower end to settle the aluminium oxide. | |||||||||||||||||||||||||||||||||||||||||||||||
5.3.2 Sample purification, | |||||||||||||||||||||||||||||||||||||||||||||||
Transfer onto the column by pipette 5.0 ml of the sample extract prepared in (5.2). Rest the pipette tip close to the column wall and allow the solution to be absorbed onto the aluminium oxide. Elute the robenidine from the column using 100 ml methanol (3.1), at a flow rate of 2 to 3 ml/minute and collect the eluate in a 250 ml round bottomed flask. Evaporate the methanol solution to dryness under reduced pressure at 40 °C by means of a rotory film evaporator (4.3). Re-dissolve the residue in 3 to 4 ml of mobile phase (3.8) and transfer quantitatively to a 10 ml graduated flask. Rinse the flask with several 1 to 2 ml portions of mobile phase and transfer these rinsings to the graduated flask. Make up to the mark with the same solvent and mix. An aliquot is filtered through a 0.45 µm (4.7). Reserve this solution for HPLC determination (5.4). | |||||||||||||||||||||||||||||||||||||||||||||||
5.4 HPLC determination. | |||||||||||||||||||||||||||||||||||||||||||||||
5.4.1 Parameters. | |||||||||||||||||||||||||||||||||||||||||||||||
The following conditions are offered for guidance, other conditions may be used provided they yield equivalent results: | |||||||||||||||||||||||||||||||||||||||||||||||
Liquid chromatographic column (4.4.1), | |||||||||||||||||||||||||||||||||||||||||||||||
HPLC mobile phase (3.8), | |||||||||||||||||||||||||||||||||||||||||||||||
Flow rate: 1.5 to 2 ml/minute, | |||||||||||||||||||||||||||||||||||||||||||||||
Detector wavelength: 317 nm, | |||||||||||||||||||||||||||||||||||||||||||||||
Injection volume: 20 to 50µl. | |||||||||||||||||||||||||||||||||||||||||||||||
Check the stability of the chromatographic system by injecting the calibration solution prepared in (3.9.3) and which contains 3.6 µg/ml, several times until constant peak heights and retention times are achieved. | |||||||||||||||||||||||||||||||||||||||||||||||
5.4.2 Calibration graph. | |||||||||||||||||||||||||||||||||||||||||||||||
Inject each of the calibration solutions, prepared in (3.9.3), several times and measure the peak heights (or areas) for each concentration. Plot a calibration curve using the mean peak heights or areas of the calibration solutions as the ordinates and corresponding concentrations in fig per ml as abscissae. | |||||||||||||||||||||||||||||||||||||||||||||||
5.4.3 Sample solution. | |||||||||||||||||||||||||||||||||||||||||||||||
Inject the sample extract (5.3.2) several times, using the same volume as taken for the calibration solutions and determine the mean peak height (area) of the robenidine peaks. | |||||||||||||||||||||||||||||||||||||||||||||||
6. Calculation of results | |||||||||||||||||||||||||||||||||||||||||||||||
From the mean height (area) of the robenidine peaks of the sample solution determine the concentration of the sample solution in µg/ml by reference to the calibration graph (5.4.2). | |||||||||||||||||||||||||||||||||||||||||||||||
The content of robenidine w (mg/kg) in the sample is given by the following formula: | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
in which: | |||||||||||||||||||||||||||||||||||||||||||||||
—c = robenidine concentration of the sample solution in µg/ml, | |||||||||||||||||||||||||||||||||||||||||||||||
—m = mass of the test position in grams. | |||||||||||||||||||||||||||||||||||||||||||||||
7. Validation of the results | |||||||||||||||||||||||||||||||||||||||||||||||
Identity. | |||||||||||||||||||||||||||||||||||||||||||||||
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract and the calibration solution (3.9.3), which contains 6 µg/ml, are compared. | |||||||||||||||||||||||||||||||||||||||||||||||
7.1.1 Co-chromatography. | |||||||||||||||||||||||||||||||||||||||||||||||
A sample extract is fortified by addition of an appropriate amount of calibration solution (3.9.3). The amount of added robenidine should be similar to the estimated amount of robenidine found in the sample extract. | |||||||||||||||||||||||||||||||||||||||||||||||
Only the height of the robenidine peak should be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its maximum height, must be within approximately 10% of the original width. | |||||||||||||||||||||||||||||||||||||||||||||||
7.1.2 Diode-array detection. | |||||||||||||||||||||||||||||||||||||||||||||||
The results are evaluated according to the following criteria: | |||||||||||||||||||||||||||||||||||||||||||||||
( a ) the wavelength of maximum absorption of the sample and of the standard spectra, recorded at the peak apex on the chromatogram, must be the same within a margin determined by the resolving power of the detection system. For diode-array detection, this is typically within approximately 2 nm; | |||||||||||||||||||||||||||||||||||||||||||||||
( b ) between 250 and 400 nm, the sample and standard spectra recorded at the peak apex on the chromatogram, must not be different for those parts of the spectrum within the range 10 to 100% of relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15% of the absorbance of the standard analyte; | |||||||||||||||||||||||||||||||||||||||||||||||
( c ) between 250 and 400 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 to 100% of relative absorbance. This criterion is met when the same maxima are present and when at all observed points the deviation between the spectra does not exceed 15% of the absorbance of the spectrum of the apex. | |||||||||||||||||||||||||||||||||||||||||||||||
If one of these criteria is not met the presence of the analyte has not been confirmed. | |||||||||||||||||||||||||||||||||||||||||||||||
7.2 Repeatability. | |||||||||||||||||||||||||||||||||||||||||||||||
The difference between the results of two parallel determinations carried out on the same sample must not exceed 10% of the higher result for robenidine content higher than 15 mg/kg. | |||||||||||||||||||||||||||||||||||||||||||||||
7.3 Recovery. | |||||||||||||||||||||||||||||||||||||||||||||||
For a fortified blank sample the recovery should be at least 85%. | |||||||||||||||||||||||||||||||||||||||||||||||
8. Results of a collaborative study | |||||||||||||||||||||||||||||||||||||||||||||||
A collaborative study was arranged by the Community in which four samples of poultry and rabbit feed, in meal or pelleted form were analysed by 12 laboratories. Duplicate analyses were performed on each sample. The results are given in the table below: | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
Sr = standard deviation of repeatability. | |||||||||||||||||||||||||||||||||||||||||||||||
CVr = coefficient of variation of repeatability. | |||||||||||||||||||||||||||||||||||||||||||||||
SR = standard deviation of reproducibility. | |||||||||||||||||||||||||||||||||||||||||||||||
CVR = coefficient of variation of reproducibility. | |||||||||||||||||||||||||||||||||||||||||||||||
33. Determination of methylbenzoquate | |||||||||||||||||||||||||||||||||||||||||||||||
7—benzyloxy-6-butyl-3-methoxycarbonyl-4-quinolone | |||||||||||||||||||||||||||||||||||||||||||||||
1. Purpose and Scope | |||||||||||||||||||||||||||||||||||||||||||||||
To determine the content of methylbenzoquate in feedingstuffs. The lower limit of determination is 1 mg/kg. | |||||||||||||||||||||||||||||||||||||||||||||||
2. Principle | |||||||||||||||||||||||||||||||||||||||||||||||
Methylbenzoquate is extracted from the sample with methanolic methanesulfonic acid solution. The extract is purified with dichloromethane, by ion-exchange chromatography and then again with dichloromethane. The methylbenzoquate content is determined by reversed-phase high-performance liquid chromatography (HPLC) with an UV detector. | |||||||||||||||||||||||||||||||||||||||||||||||
3. Reagents | |||||||||||||||||||||||||||||||||||||||||||||||
3.1 Dichloromethane. | |||||||||||||||||||||||||||||||||||||||||||||||
3.2 Methanol, HPLC grade. | |||||||||||||||||||||||||||||||||||||||||||||||
3.3 HPLC mobile phase: | |||||||||||||||||||||||||||||||||||||||||||||||
mixture of methanol (3.2) and water (HPLC grade) 75+25 (v+v). | |||||||||||||||||||||||||||||||||||||||||||||||
Filter through a 0.22 µm filter (4.5) and degas the solution (e.g. by ultrasonification for 10 minutes). | |||||||||||||||||||||||||||||||||||||||||||||||
3.4 Methanesulfonic acid solution, ó = 2%. | |||||||||||||||||||||||||||||||||||||||||||||||
Dilute 20.0 ml methanesulfonic acid to 1000 ml with methanol (3.2). | |||||||||||||||||||||||||||||||||||||||||||||||
3.5 Hydrochloric acid solutions, ó = 10%. | |||||||||||||||||||||||||||||||||||||||||||||||
Dilute 100 ml hydrochloric acid (p20 c. 1.18 g/ml) to 1000 ml with water. | |||||||||||||||||||||||||||||||||||||||||||||||
3.6 Cation-exchange resin Amberlite CG-120 (Na), 100 to 200 mesh | |||||||||||||||||||||||||||||||||||||||||||||||
The resin is pretreated before use: slurry 100 g resin with 500 ml hydrochloric acid solution (3.5) and heat on a hot plate to boiling, stirring continuously. Allow to cool and decant off the acid. Filter through a filter paper under vacuum. Wash the resin twice with 500 ml portions of water and then with 250 ml of methanol (3.2). Rinse the resin with a further 250 ml portion of methanol and dry by passing air through the filter cake. Store the dried resin in a stoppered bottle. | |||||||||||||||||||||||||||||||||||||||||||||||
3.7 Standard substance: pure methyl benzoquate(7—benzyloxy-6-butyl-3-methoxycarbonyl-4-quinolone). | |||||||||||||||||||||||||||||||||||||||||||||||
3.7.1 Methyl benzoquate stock standard solution, 500 µg/ml. | |||||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest 0.1 mg, 50 mg of standard substance (3.7), dissolve in methanesulfonic acid solution (3.4) in a 100 ml graduated flask, make up to the mark and mix. | |||||||||||||||||||||||||||||||||||||||||||||||
3.7.2 Methylbenzoquate intermediate standard solution, 50 µg/ml. | |||||||||||||||||||||||||||||||||||||||||||||||
Transfer 5.0 ml of methylbenzoquate stock standard solution (3.7.1) into a 50 ml graduated flask, make up the mark with methanol (3.2) and mix. | |||||||||||||||||||||||||||||||||||||||||||||||
3.7.3 Calibration solutions. | |||||||||||||||||||||||||||||||||||||||||||||||
Transfer 1.0, 2.0, 3.0, 4.0, and 5.0 ml of mthylbenzoquate intermediate standard solution (3.7.2) into a series of 25 ml graduated flasks. Make up to the mark with the mobile phase (3.3) and mix. These solutions have concentrations of 2.0, 4.0, 6.0, 8.0, and 10.0 µg/ml methylbenzoquate respectively. These solutions must be freshly prepared before use. | |||||||||||||||||||||||||||||||||||||||||||||||
4. Apparatus | |||||||||||||||||||||||||||||||||||||||||||||||
4.1 Laboratory shaker. | |||||||||||||||||||||||||||||||||||||||||||||||
4.2 Rotary film evaporator. | |||||||||||||||||||||||||||||||||||||||||||||||
4.3 Glass column (250 mm X 15 mm) fitted with a stopcock and reservoir of approximately 200 ml capacity. | |||||||||||||||||||||||||||||||||||||||||||||||
HPLC equipment with variable wavelength ultraviolet detector or diode-array detector. | |||||||||||||||||||||||||||||||||||||||||||||||
4.4.1 Liquid chromatographic column: 300 mm x 4 mm, C-18, 10 µm packing or equivalent. | |||||||||||||||||||||||||||||||||||||||||||||||
4.5 Membrane filters, 0.22 µm. | |||||||||||||||||||||||||||||||||||||||||||||||
4.5 Membrane filters, 0.45 µm. | |||||||||||||||||||||||||||||||||||||||||||||||
5. Procedure | |||||||||||||||||||||||||||||||||||||||||||||||
5.1 General. | |||||||||||||||||||||||||||||||||||||||||||||||
5.1.1 A blank feed should be analysed to check that neither methyl-benzoquate nor interfering substances are present. | |||||||||||||||||||||||||||||||||||||||||||||||
5.1.2 A recovery test should be carried out by an analysing the blank feed which has been fortified by addition of a quantity of methylbenzoquate, similar to that present in the sample. To fortify at a level of 15 mg/kg, and 600 µl of the stock standard solution (3.7.1) to 20 g of the blank feed, mix and wait for 10 minutes before proceeding with the extraction step (5.2). | |||||||||||||||||||||||||||||||||||||||||||||||
Note: For the purpose of this method, the blank feed should be similar in type to that of the sample and on analysis methylbenzoquate should not be detected. | |||||||||||||||||||||||||||||||||||||||||||||||
5.2 Extraction. | |||||||||||||||||||||||||||||||||||||||||||||||
Weigh to the nearest 0.01 g, approximately 20 g of the prepared sample and transfer to a 250 ml conical flask. Add 100.0 ml of methanesulfonic acid solution (3.4) and shake mechanically (4.1) for 30 minutes. Filter the solution through a filter paper and retain the filtrate for the liquid-liquid partition step (5.3). | |||||||||||||||||||||||||||||||||||||||||||||||
5.3 Liquid-liquid partition. | |||||||||||||||||||||||||||||||||||||||||||||||
Transfer into a 500 ml separating funnel containing 100 ml of hydrochloric acid solution (3.5), 250 ml of the filtrate obtained in (5.2). Add 100 ml dichloromethane (3.1) to the funnel and shake for one minute. Allow the layers to separate and run off the lower (dichloromethane) layer into a 500 ml round-bottomed flask. Repeat the extraction of the aqueous phase with two further 40—ml portions of dichloromethane and combine these with the first extract in the round-bottomed flask. Evaporate the dichloromethane extract down to dryness on the rotary evaporator (4.2) operating under reduced pressure at 40 °C. Dissolve the residue in 20 to 25 ml methanol (3.2), stopper the flask and retain the whole of the extract for ion-exchange chromatography (5.4). | |||||||||||||||||||||||||||||||||||||||||||||||
5.4 Ion-exchange chromatography. Preparation of the cation-exchange column. | |||||||||||||||||||||||||||||||||||||||||||||||
Insert a plug of glass wool into the lower end of a glass column (4.3). Prepare a slurry of 5.0 g of the treated cation-exchange resin (3.6) with 50 ml of hydrochloric acid (3.5), pour into the glass column and allow to settle. Run out the excess acid to just above the resin surface and wash the column with water until the effluent is neutral to litmus. Transfer 50 ml methanol (3.2) onto the column and allow to drain down to the resin surface. | |||||||||||||||||||||||||||||||||||||||||||||||
5.4.2 Column chromatography. | |||||||||||||||||||||||||||||||||||||||||||||||
By means of a pipette, carefully transfer the extract obtained in (5.3) onto the column. Rinse the round-bottomed flask with two portions of 5 to 10 ml methanol (3.2) and transfer these washings to the column. Run the extract down to the resin surface and wash the column with 50 ml methanol, ensuring that the flow rate does not exceed 5 ml per minute. Discard the effluent. Elute the methyl benzoquate from the column using 150 ml of methanesulfonic acid solution (3.4) and collect the column eluate in a 250 ml conical flask. | |||||||||||||||||||||||||||||||||||||||||||||||
5.5 Liquid-liquid partition. | |||||||||||||||||||||||||||||||||||||||||||||||
Transfer the eluate obtained in (5.4.2) into a 1 litre separating funnel. Rinse the conical flask with 5 to 10 ml methanol (3.2) and combine the washings with the contents of the separating funnel. Add 300 ml of hydrochloric acid solution (3.5) and 130 ml of dichloremethane (3.1). Shake for 1 minute and allow the phases to separate. Run off the lower (dichloromethane) layer into a 500 ml round-bottomed flask. Repeat the extraction of the aqueous phase with two further 70 ml portions of dichloromethane and combine these extracts with the first in the round-bottomed flask. | |||||||||||||||||||||||||||||||||||||||||||||||
Evaporate the dichloromethane extract down to dryness on the rotary evaporator (4.2) operating under reduced pressure at 40°C. Dissolve the residue in the flask with approximately 5 ml of methanol (3.2) and transfer this solution quantitatively to a 10 ml graduated flask. Rinse the round-bottomed flask with a further two portions of 1 to 2 ml of methanol and transfer these to the graduated flask. Make up to the mark with methanol and mix. An aliquot portion is filtered through a membrane filter (4.6). Reserve this solution for HPLC-determination (5.6). | |||||||||||||||||||||||||||||||||||||||||||||||
5.6 HPLC determination. | |||||||||||||||||||||||||||||||||||||||||||||||
5.6.1 Parameters. | |||||||||||||||||||||||||||||||||||||||||||||||
The following conditions are offered for guidance, other conditions may be used provided that they give equivalent results: | |||||||||||||||||||||||||||||||||||||||||||||||
— liquid chromatographic column (4.4.1), | |||||||||||||||||||||||||||||||||||||||||||||||
— HPLC mobile phase; methanol water mixture (3.3), | |||||||||||||||||||||||||||||||||||||||||||||||
— flow rate: 1 to 1.5 ml/minute, | |||||||||||||||||||||||||||||||||||||||||||||||
— detection wavelength: 265 nm, | |||||||||||||||||||||||||||||||||||||||||||||||
— injection volume: 20 to 50 µl. | |||||||||||||||||||||||||||||||||||||||||||||||
Check the stability of the chromatographic system, by injecting the calibration solution prepared in (3.7.3) and which contains 4 µg/ml, several times until constant peak heights or areas and retention times are achieved. | |||||||||||||||||||||||||||||||||||||||||||||||
5.6.2 Calibration graph. | |||||||||||||||||||||||||||||||||||||||||||||||
Inject each of the calibration solutions, prepared in (3.7.3), several times and measure the peak heights (or areas) for each concentration. Plot a calibration graph using the mean peak heights (or areas) of the calibration solutions as the ordinates and the corresponding concentrations in µg/ml as the abscissae. | |||||||||||||||||||||||||||||||||||||||||||||||
5.6.3 Sample solution. | |||||||||||||||||||||||||||||||||||||||||||||||
Inject the sample extract (5.5) several times, using the same volume as taken for the calibration solutions and determine the mean peak height (area) of the methylbenzoquate peaks. | |||||||||||||||||||||||||||||||||||||||||||||||
6. Calculation of results | |||||||||||||||||||||||||||||||||||||||||||||||
Determine the concentration of the sample solution in µg/ml from the mean height (area) of the methylbenzoquate peaks of the sample solution by reference to the calibration graph (5.6.2). | |||||||||||||||||||||||||||||||||||||||||||||||
The content of methylbenzoquate w (mg/kg) of the sample is given by the following formula: | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
in which: | |||||||||||||||||||||||||||||||||||||||||||||||
c = methylbenzoquate concentration of the sample solution in µg/ml. | |||||||||||||||||||||||||||||||||||||||||||||||
m = mass of the test portion in grams. | |||||||||||||||||||||||||||||||||||||||||||||||
7. Validation of the results | |||||||||||||||||||||||||||||||||||||||||||||||
7.1 Identity. | |||||||||||||||||||||||||||||||||||||||||||||||
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract and the calibration solution (3.7.3) which contains 10 µg/ml are compared. | |||||||||||||||||||||||||||||||||||||||||||||||
7.1.1 Co-chromatography. | |||||||||||||||||||||||||||||||||||||||||||||||
A sample extract is fortified by addition of an appropriate amount of the intermediate standard solution (3.7.2). The amount of added methylbenzoquate should be similar to the estimated amount of methylbenzoquate in the sample extract. | |||||||||||||||||||||||||||||||||||||||||||||||
Only the height of the methylbenzoquate peak should be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its maximum height, must be within 10% of the original width. | |||||||||||||||||||||||||||||||||||||||||||||||
7.1.2 Diode-array detection. | |||||||||||||||||||||||||||||||||||||||||||||||
The results are evaluated according to the following criteria: | |||||||||||||||||||||||||||||||||||||||||||||||
( a ) the wavelength of maximum absorption of the sample and of the standard spectra recorded at the peak apex on the chromatogram must be the same within a margin determined by the resolving power of the detection system. For diode-array detection, this is typically within approximately 2 nm; | |||||||||||||||||||||||||||||||||||||||||||||||
( b ) between 220 and 350 nm, the sample and standard spectra recorded at the peak apex on the chromatogram, must not be different for those parts of the spectrum within the range 10 to 100% of relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15% of the absorbance of the standard analyte; | |||||||||||||||||||||||||||||||||||||||||||||||
( c ) between 220 and 350 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 to 100% of relative absorbance. This criterion is met when the same maxima are present and when at all observed points the deviation between the spectra does not exceed 15% of the absorbance of the spectrum of the apex. | |||||||||||||||||||||||||||||||||||||||||||||||
If one of these criteria is not met the presence of the analyte has not been confirmed. | |||||||||||||||||||||||||||||||||||||||||||||||
7.2 Repeatability. | |||||||||||||||||||||||||||||||||||||||||||||||
The difference between results of two parallel determinations carried out on the same sample must not exceed 10%, relative to the higher result, for methylbenzoquate contents between 4 and 20 mg/kg. | |||||||||||||||||||||||||||||||||||||||||||||||
7.3 Recovery. | |||||||||||||||||||||||||||||||||||||||||||||||
For a fortified blank sample the recovery should be at least 90%. | |||||||||||||||||||||||||||||||||||||||||||||||
8. Results of a collaborative study | |||||||||||||||||||||||||||||||||||||||||||||||
Five samples were analysed by 10 laboratories. Duplicate analyses were performed on each sample. The results are given in the Table below: | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
n.d.: not detected. | |||||||||||||||||||||||||||||||||||||||||||||||
Sr: standard deviation of repeatability. | |||||||||||||||||||||||||||||||||||||||||||||||
CVr: coefficient of variation of repeatability. | |||||||||||||||||||||||||||||||||||||||||||||||
SR: standard deviation of reproducibility. | |||||||||||||||||||||||||||||||||||||||||||||||
CVR: coefficient of variation of reproducibility. | |||||||||||||||||||||||||||||||||||||||||||||||
GIVEN under my Official Seal, this 1st day of September, 1994. | |||||||||||||||||||||||||||||||||||||||||||||||
JOE WALSH, | |||||||||||||||||||||||||||||||||||||||||||||||
Minister for Agriculture, Food | |||||||||||||||||||||||||||||||||||||||||||||||
and Forestry. | |||||||||||||||||||||||||||||||||||||||||||||||
EXPLANATORY NOTE. | |||||||||||||||||||||||||||||||||||||||||||||||
These Regulations amend the European Communities (Feedingstuffs) (Method of Analysis) Regulations, 1978, ( S.I. No. 250 of 1978 ) so as to give effect to:— | |||||||||||||||||||||||||||||||||||||||||||||||
(1) the Eleventh Commission Directive 93/70/EEC which establishes a Community method of analysis for the medicinal additive halofuginone in feedingstuffs; | |||||||||||||||||||||||||||||||||||||||||||||||
(2) the Twelfth Commission Directive 93/117/EEC which establishes Community methods of analysis for the medicinal additives robenidine and methylbenzoquate in feedingstuffs. | |||||||||||||||||||||||||||||||||||||||||||||||