| |
S.I. No. 139 of 1994.
|
| |
EUROPEAN COMMUNITIES (AUTHORIZATION, PLACING ON THE MARKET, USE AND CONTROL OF PLANT PROTECTION PRODUCTS) REGULATIONS, 1994.
|
| |
I, JOE WALSH, Minister for Agriculture, Food and Forestry, in exercise of the powers conferred on me by
section 3
of the
European Communities Act, 1972
(No. 27 of 1972), and for the purpose of giving effect to Council Directive No 91/414/EEC of 15 July 19911, the Corrigendum to Council Directive No. 91/414/EEC , Commission Regulation (EEC) No 3600 of 11 December 19923 ,and Commission Directive No 93/71/EEC of 27 July 19934, taking account of Council Directive No. 78/631/EEC of 26 June 19785, Council Directive 81/187/EEC of 26 March 19816, and Commission Directive 84/291/EEC of 18 April 19847, and further taking account of Council Directive 67/548/EEC of 27 June 19678, as amended by Council Directive 92/32/EEC of 30 April 1992 9, hereby make the following Regulations:
|
|
|
1 ..
|
1. (1) These Regulations may be cited as the European Communities (Authorization, Placing on the Market, Use and Control of Plant Protection Products) Regulations, 1994.
|
| |
(2) These Regulations shall come into operation on the first day of October 1994.
|
|
|
2 Interpretation.
|
2. (1) In these Regulations—
|
| |
"Annex I" means Annex I to the Directive of 1991, as amended;
|
| |
"Annex II" (which is set out in Part 1 of the First Schedule) means Annex II to the Directive of 1991, as amended by Commission Directive No 93/71/EEC of 27 July 1993;
|
| |
1O.J. No L230/1 19/8/1991
|
| |
2 O.J. No L170/40 25/6/1992
|
| |
3 O.J. No L366/10 15/12/1992
|
| |
4 O.J. No L221/27 31/8/93
|
| |
5 O.J. No L206/13 29/7/1978
|
| |
6 O.J. No L 88/29 2/4/1981
|
| |
7 O.J. No L144/1 30/5/1984
|
| |
8 O.J. No L196/1 16/8/1967
|
| |
9 O.J. No L154/1 5/6/1992
|
| |
"Annex III" (which is set out in Part 2 of the First Schedule) means Annex III to the Directive of 1991, as amended by Commission Directive No 93/71/EEC of 27 July 1993;
|
| |
"Annex IV" (which is set out in Part 3 of the First Schedule) means Annex IV to the Directive of 1991;
|
| |
"Annex V" (which is set out in Part 4 of the First Schedule) means Annex V to the Directive of 1991;
|
| |
"Annex VI" (which is set out in Part 5 of the First Schedule) means Annex VI to the Directive of 1991;
|
| |
"Annex VII" (which is set out in Part 6 of the First Schedule) means Annex IX to the Directive of 1967 as amended by the Directive of 1992, and adapted by Commission Directive No 91/410/EEC10;
|
| |
"Annex VIII" (which is set out in Part 7 of the First Schedule) means Annex II to the Directive of 1967 as amended by the Directive of 1992, and adapted by Commission Directive No. 93/21/EEC11;
|
| |
"Annex IX" (which is set out in Part 8 of the First Schedule) means Annex III to the Directive of 1967 as amended by the Directive of 1992, and adapted by Commission Directive No. 93/21/EEC;
|
| |
"Annex X" (which is set out in Part 9 of the First Schedule) means Annex V to the Directive of 1978;
|
| |
"Annex XI" (which is set out in Part 10 of the First Schedule) means Annex IV to the Directive of 1967 as amended by the Directive of 1992, and adapted by Commission Directive No. 93/21/EEC;
|
| |
"Annex XII" means Annex XII set out in Part 11 of the First Schedule, which comprises additional safety advice in accordance with Article 16 (5) of the Directive of the Directive of 1991;
|
| |
"aircraft" includes hovercraft;.
|
| |
"authorised officer" means an officer of the Minister appointed in writing by the Minister to be an authorised officer for the purpose of these Regulations;
|
| |
10O.J. No. L228/68, 17/8/1991.
|
| |
11 O.J. No. L110/20, 4/5/1993.
|
| |
"the Commission" means the Commission of the European Communities;
|
| |
"the competent authority" for the purposes of these Regulations and Commission Regulation No. 3600 of 11 December 1992 is the Pesticide Control Service of the Department of Agriculture, Food and Forestry;
|
| |
"controlled product" means a product specified by the Minister as such through Orders made pursuant to Regulation 28;
|
| |
"designated chemist" means an officer of the Minister holding the position of a chemist authorized in writing by the Minister for the purposes of these Regulations;
|
| |
"the Directive of 1991" means Council Directive No. 91/414/EEC of 15 July 19911;
|
| |
"the Directive of 1967" means Council Directive 67/548/EEC of 27 June, 19678;
|
| |
"the Directive of 1978" means Council Directive No. 78/631/EEC of 26 June, 19785, as amended and adapted;
|
| |
"the Directive of 1992" means Council Directive No. 92/32/EEC of 30 April, 19929, as amended and adapted;
|
| |
"good agricultural practice" in the use of a plant protection product, means safe use of the plant protection product under actual conditions necessary for its effective action, in accordance with an authorization granted pursuant to these Regulations, encompassing a range of levels of application up to the highest level of use for which an authorization has been granted, applied in a manner which leaves a residue which is the smallest practicable; safe use, in relation to good agricultural practice, means taking into account public and occupational health and environmental considerations; actual conditions of use include any stage in the production, storage, transport and distribution of plants and plant products;
|
| |
"good plant protection practice" in the use of plant protection products, means their responsible use in accordance with principles defining such use, as set out in the Second Schedule hereto, taking account of the range of authorized products available, the spectrum of harmful organisms occurring, the production, storage, transport and distribution conditions for plants and plant products and alternative means of plant protection available;
|
| |
1 O.J. No. L230/1 19/8/1991
|
| |
8 O.J. No. L196/1 16/8/1967
|
| |
5 O.J. No. L206/13 29/7/1978
|
| |
9 O.J. No. L154/1 5/6/1992
|
| |
"Member State" means a Member State of European Communities;
|
| |
"the Minister" means the Minister for Agriculture, Food and Forestry;
|
| |
"officially recognized testing facilities and organizations" for the purposes of these Regulations, means testing facilities and organizations which carry out experiments, studies, tests and analyses in accordance with these Regulations;
|
| |
"officially recognized tests and analyses" for the purposes of these Regulations, means experiments, studies, tests and analyses carried out in accordance with methodologies and to a standard specified from time to time by the competent authority and issued as guideline documentation;
|
| |
"the Regulations of 1994" means the European Communities (Classification, Packaging and Labelling of Pesticides) Regulations, 1994 (
S.I. No. 138 of 1994
);
|
| |
"the State Chemist" means the Head of the State Laboratory or a member of the staff of the State Laboratory holding the position of a chemist authorized by the State Chemist in writing to perform functions assigned to the State Chemist under Regulation 34;
|
| |
"trials permit" means a permit granted under Regulation 26.
|
| |
(2) In these Regulations, unless otherwise indicated—
|
| |
( a ) a reference to a Regulation is a reference to a Regulation of these Regulations,
|
| |
( b ) a reference to a paragraph or subparagraph is a reference to a paragraph or subparagraph of the provision in which the reference occurs,
|
| |
( c ) a reference to a Schedule is a reference to a Schedule to these Regulations.
|
| |
(3) A word or expression that is used in the Directive of 1991 or in any other Council or Commission Directive of the European Communities mentioned in these Regulations has, unless the contrary intention appears, the meaning in these Regulations that it has in the Directive concerned.
|
|
|
3 Application
|
3. These Regulations apply to any plant protection product which is a plant protection product for the purposes of the Directive of 1991.
|
|
|
4 ..
|
4. (1) Subject to paragraphs (2) and (3), the placing on the market and use of a plant protection product in the form in which it is supplied to the user and intended for use as such is hereby prohibited unless the requirements of these Regulations regarding its authorization are complied with.
|
| |
(2) The placing on the market of a plant protection product in the form in which it is supplied to the user and intended for use as such is hereby prohibited if—
|
| |
( a ) the net quantity in any container of such a plant protection product is less than the quantity stated there on in the manner specified in Article 16(1) (d) of the Directive of 1991, or
|
| |
( b ) the fastenings or containers used to package the plant protection product have been tampered with.
|
| |
(3) The placing on the market of a plant protection product in the form in which it is supplied to the user and intended for use as such is hereby prohibited unless the annual fees as provided for in Regulation 36 (4) have been paid by the dates specified in accordance with the provisions of that Regulation.
|
| |
(4) These Regulations shall not apply to—
|
| |
( a ) the production, storage or movement of a plant protection product which has not been authorized, where it is intended for use in another Member State and where—
|
| |
(i) the product is authorized in the other Member State, and
|
| |
(ii) the provisions of Regulation 30 are complied with.
|
| |
(5) Regulations 6, 7, 8, 12, 13, 15, 16 and 18 shall not apply to plant protection products authorized for trials purposes in accordance with Regulations 25 and 26.
|
|
|
5 Exemptions from Certain Provisions of the European Communities (Classification, Packaging and Labelling of Pesticides) Regulations, 1994 (
S.I. No 138 of 1994
) and from certain provisions of these Regulations
|
5. (1) Plant protection products which are pesticides referred to in paragraph (1) (a) of Regulation 3 of the Regulations of 1994, which are classified in accordance with Regulation 5 of those Regulations—
|
| |
( a ) are hereby exempted from the provisions of Regulation 6 paragraphs (2) (a) and (b) and Regulation 18 of those Regulations, and
|
| |
( b ) are hereby deemed to comply with the provisions of Regulation 6 paragraph (1) and Regulations 7 and 8 of the Regulations of 1994,
|
| |
where they have been authorized in accordance with these Regulations.
|
| |
(2) Plant protection products which are pesticides referred to in paragraph (1) (a) of Regulation 3 of the Regulations of 1994 and which are on the market before these Regulations come into effect, may continue to be placed on the market for use in accordance with the Regulations of 1994, until such time as they are authorized in accordance with these Regulations, or are refused such authorization.
|
| |
(3) Notwithstanding the provisions of Regulations 8, 13 and 18, in the case of a plant protection product which is not on the market before these Regulations come into effect, where the plant protection product is similar to and considered by the competent authority to involve no greater risk for man, animals or for the environment than a plant protection product placed on the market in accordance with the provisions of the Regulations of 1994 and—
|
| |
( a ) the record of the studies conducted and the information, documentation and materials referred to in Regulation 6 (2) (a) of those Regulations, has not yet been approved, or
|
| |
( b ) at least one active substance contained in the plant protection product is contained in a plant protection product cleared in accordance with those Regulations,
|
| |
permission may be granted by the competent authority on approval of an accurate record of a limited number of studies, information, supporting documentation and materials, as specified from time to time by the competent authority, to market and use such a plant protection product, where application is made in the form set out in the Second Schedule, unless in the case of the plant protection product already on the market, in accordance with Regulation 10, the periods specified in that Regulation have not yet expired for information referred to in Regulation 8 (3) (a) and (b).
|
|
|
6 Use of Plant Protection Products
|
6. (1) The use of a plant protection product other than as specified in paragraph (2), is hereby prohibited.
|
| |
(2) Plant protection products shall be used—
|
| |
( a ) in compliance with the conditions established in accordance with Regulations 13, 15, 18 and 19 and specified on the labelling, as appropriate,
|
| |
( b ) in compliance with the conditions established in accordance with Regulation 16, as appropriate,
|
| |
( c ) in the case of plant protection products on the market before these Regulations come into effect, 1 October 1994 and pending their authorization in accordance with these Regulations, in compliance with the conditions specified on the labelling, in accordance with the requirements of the Regulations of 1994,
|
| |
( d ) in accordance with the Principles of Good Plant Protection Practice as set out in the Third Schedule, and
|
| |
( e ) where possible, in accordance with the principles of integrated control.
|
|
|
7 Marketing of Active Substances
|
7. (1) Subject to paragraph (2), the placing on the market of active substances is hereby prohibited unless—
|
| |
( a ) they are classified, packaged and labelled in accordance with the Directive of 1967, and
|
| |
( b ) where the active substance was not on the market 2 years after notification of the Directive of 1991, a dossier has been forwarded to the Member States and the Commission, in accordance with Article 6 of that Directive, with the declaration that the active substance is intended for a use specified in Article 2 (1) of that same Directive.
|
| |
(2) This Regulation shall not apply to active substances contained in plant protection products intended for use for trials purposes in accordance with Regulations 25 and 26.
|
|
|
8 Application for Authorization
|
8. (1) Every application for authorization of a plant protection product intended to be placed on the market, shall be made by or on behalf of the person responsible for first placing it on the market. Every applicant for an authorization shall have a business premises in a Member State. Applications shall be in the form set out in Part 1 of the Fourth Schedule.
|
| |
(2) Applications, which shall be supported with the documentation specified in paragraph (3) and where relevant, paragraphs (5) (b) and (6), shall be in the English language and shall be in the form set out in Part 2 of the Fourth Schedule.
|
| |
(3) Every application for authorization of a plant protection product, shall be submitted to the competent authority and shall be supported with—
|
| |
( a ) a dossier satisfying, in the light of current scientific and technical knowledge, the requirements set out in Annex III, and
|
| |
( b ) for each active substance in the plant protection product, a dossier satisfying, in the light of current scientific and technical knowledge, the requirements set out in Annex II,
|
| |
( c ) samples of the packaging and models, drafts or samples of labelling and leaflets referred to in Regulations 23 and 24,
|
| |
( d ) samples of the plant protection product and of each active substance included in it and analytical standards for each such active substance, for impurities and formulants of toxicological or environmental significance, and for transformation products of the active substance included in the residue definition, and
|
| |
( e ) the relevant fee in accordance with Regulation 36.
|
| |
(4) The tests and analyses conducted for the purposes of compiling dossiers referred to in paragraph (3) (a), shall be carried out under agricultural, plant health and environmental conditions relevant to use of the plant protection product in question and representative of those prevailing where the product is intended to be used, and shall be officially recognized tests and analyses.
|
| |
(5) Notwithstanding paragraph (2), and subject to Regulation 10—
|
| |
( a ) applicants shall be exempted from supplying the information required under paragraph 3 (b), except for that identifying the active substance, if the active substance is already listed in Annex I, taking into account the conditions of inclusion in Annex I, and does not differ significantly in degree of purity and nature of impurities, from the composition registered in the Annex II dossier accompanying the original application, and
|
| |
( b ) in the case of a plant protection product already authorized in another Member State, at the request of an applicant, who must substantiate the claim to comparability with documentary evidence, applicants shall be exempted from repeating tests and analyses already carried out in connection with the authorization of the product, to the extent that agricultural, plant health and environmental (including climatic) conditions relevant to the use of the product are comparable in the regions concerned.
|
| |
(6) Notwithstanding paragraph (2), and subject to Regulation 10, every application for authorization of a plant protection product in accordance with Regulation 13, (2), taking account of the agricultural, plant health and environmental (including climatic) conditions relevant to the use of the product in the regions concerned, shall be supported with a claim as to the comparability of the regions concerned and shall be supported with documentary evidence to support any such claim.
|
|
|
9 Limiting of Testing involving Vertebrate Species
|
9. (1) Notwithstanding the provisions of Regulations 8 and 10, applicants for authorization of plant protection products shall, before carrying out experiments involving vertebrate animals, enquire of the competent authority—
|
| |
( a ) whether the plant protection product for which an application is to be made is the same as a plant protection product for which authorization has been granted, and
|
| |
( b ) as to the name and address of the holder of the authorization.
|
| |
(2) Where the competent authority is satisfied on the basis of documentary evidence provided that the prospective applicant intends to apply for authorization on his own behalf and that the other information specified in Regulation 8 (3) is available to him or for use on his behalf, it shall provide the name and address of any holder of a previous relevant authorization and shall at the same time inform that holder of the name and address of the applicant.
|
| |
(3) The holder of a previous authorization and the applicant shall take all reasonable steps to reach agreement on the sharing of information so as to avoid the duplication of testing on vertebrate animals.
|
| |
(4) Where data is to be submitted with a view to inclusion in Annex I of an active substance already on the market 2 years after notification of the Directive of 1991, the competent authority shall encourage applicants to cooperate in the provision of the requested data, with a view to limiting the duplication of testing on vertebrate animals.
|
| |
(5) Where application is made for the inclusion in Annex I of an active substance already on the market 2 years after notification of the Directive of 1991, applicants shall take all reasonable steps to reach agreement on—
|
| |
( a ) the sharing of relevant data and information, and
|
| |
( b ) the submission collectively of all the data and information concerned.
|
|
|
10 Data Protection
|
10. (1) Information contained in the dossier referred to in Regulation 8 (3) (b) shall not be used to the benefit of other applicants unless—
|
| |
( a ) the applicant has agreed with the first applicant that use may be made of such information and the first applicant has submitted written confirmation of such agreement, or
|
| |
( b ) 10 years have elapsed from the first inclusion in Annex I of an active substance first placed on the market in the European Communities as a constituent of a plant protection product 2 years after the notification of the Directive of 1991, or
|
| |
( c ) 10 years have elapsed from the date of first marketing within the territory of the state of an active substance as a constituent of a plant protection product which was on the market 2 years after the notification of the Directive of 1991, and
|
| |
5 years have elapsed from the date of decision in the case of further information which is generated specifically for and is necessary—
|
| |
(i) for first inclusion of an active substance in Annex I, or
|
| |
(ii) to vary the conditions for, or to maintain, the inclusion of an active substance in Annex I.
|
| |
(2) Notwithstanding the period of 5 years provided for in paragraph (1), where that period expires before the periods provided for in paragraphs (1) (b) and (1) (c) the period of 5 years shall be extended so as to expire on the same date as those periods.
|
| |
(3) Information contained in the dossier referred to in Regulation 8 (3) (a) shall not be used to the benefit of other applicants unless—
|
| |
( a ) the applicant has agreed with the first applicant that use may be made of such information and the first applicant has submitted written confirmation of such agreement, or
|
| |
( b ) 10 years have elapsed from the first authorization or marketing of the plant protection product in any Member State, where authorization follows the inclusion in Annex I of any active substance contained in the product, or
|
| |
( c ) 10 years have elapsed from the date of first marketing within the territory of the state of a plant protection product where such marketing precedes inclusion in Annex I of any active substance contained in the product.
|
| |
(4) The provisions of subparagraphs (1) (c), (2) and (3) (c) also apply to data and information submitted in accordance with the provisions of the Regulations of 1994.
|
| |
(5) The competent authority shall inform the Commission of each instance in which an application for authorization of a plant protection product is being considered where they consider an active substance to be listed in Annex I, that has been produced by a person or manufacturing process other than those specified in the dossier on the basis of which the active substance was first included in Annex I.
|
| |
(6) In each instance referred to in paragraph (5), the competent authority shall transmit to the Commission all data regarding the identity and impurities of the active substance concerned.
|
|
|
11 Confidentiality
|
11. (1) An applicant for authorization of a plant protection product, who claims that certain information submitted in accordance with Regulation 8, includes information involving industrial and commercial secrets, may make application for such information to be treated as confidential. Any such application shall identify the information concerned and shall for each such item of information include a statement justifying it being treated as confidential.
|
| |
(2) Subject to paragraphs (3) and (4) and without prejudice to Council Directive 90/313/EEC of 7 June 1990 on the freedom of access to information on the environment12, the competent authority shall ensure that information referred to in paragraph (1) is treated as confidential where it is accepted by the competent authority that such treatment is warranted.
|
| |
(3) Confidentiality shall not apply to—
|
| |
( a ) the names and content of the active substance or substances and the name of the plant protection product,
|
| |
( b ) the name of other substances which are regarded as dangerous under Directives 67/548/EEC and 78/631/EEC,
|
| |
( c ) physico-chemical data concerning the active substance and plant protection product,
|
| |
( d ) any ways of rendering the active substance or plant protection product harmless,
|
| |
12 O.J. No. L158/56 23/6/1990
|
| |
( e ) a summary of the results of the tests to establish the substance's or product's efficacy and harmlessness to humans, animals, plants and the environment,
|
| |
( f ) recommended methods and precautions to reduce handling, storage, transport, fire or other hazards,
|
| |
( g ) methods of analysis referred to in Article 4(1)(c) and (d) and 5(1) of the Directive of 1991,
|
| |
( h ) methods of disposal of the product and of its packaging,
|
| |
( i ) decontamination procedures to be followed in the case of accidental spillage or leakage, or
|
| |
( j ) first aid and medical treatment to be given in the case of injury to persons.
|
| |
(4) Where applicants subsequently disclose previously confidential information, they shall inform the competent authority accordingly.
|
|
|
12 Consideration of Applications for Authorization of Plant Protection Products and for Inclusion of Active Substances in Annex I
|
12. (1) The competent authority shall consider each application received for the authorization of a plant protection product and provided that it has the necessary scientific and technical resources at its disposal, shall inform the applicant within a reasonable period, of its decision as to whether the application has been granted or refused.
|
| |
(2) In the case of applications involving one or more active substances not on the market in the European Communities as a constituent of a plant protection product 2 years after notification of the Directive of 1991, and not subsequently included in Annex I, the competent authority shall, without undue delay, assess the information provided to determine if the requirements specified in Regulation 8 (3) have been satisfied. For each such application believed to satisfy the requirements of Annex II, the competent authority shall require the applicant to forward the dossier to the competent authorities of the other Member States and to the Commission together with a dossier complying with Annex III on at least one preparation containing that active substance.
|
| |
(3) Where in accordance with paragraph (2), applicants are required to forward a dossier believed to satisfy the requirements of Annex II and a dossier complying with Annex III on at least one preparation containing the active substance, the competent authority shall, in accordance with Article 6.3, request the Commission to establish whether the dossiers satisfy the requirements of Annex II and Annex III, in accordance with the procedure provided for in that Article.
|
|
|
13 Granting of Authorizations
|
13. (1) The competent authority shall not authorize the placing on the market and use of any plant protection product unless—
|
| |
( a ) its active substances are listed in Annex I and any conditions laid down therein are fulfilled,
|
| |
( b ) following application of the uniform principles as contained in Annex VI, the requirements of Article 4 (1) (b), (c), (d) and (e) of the Directive of 1991 are satisfied,
|
| |
( c ) maximum residue levels in the agricultural products referred to in the authorization have been provisionally established, and notified to the Commission in accordance with Regulation 22, such maximum residue levels, when adopted pursuant to paragraph (1) of Regulation 28, to remain in force until replaced in accordance with the procedures specified in Article 4 (1) (f) of the Directive of 1991, and
|
| |
( d ) its packaging and labelling satisfy the provisions of Regulations 23 and 24.
|
| |
(2) Subject to paragraphs (3) and (4), in the case of a plant protection product already authorised in accordance with the provisions of the Directive of 1991 in another Member State, and where the product contains only active substances included in Annex I, at the request of the applicant, the competent authority, to the extent that Annex VI has been adopted in accordance with Article 23 of the Directive of 1991, shall authorize the placing on the market and use of the product, where it has been established that the agricultural, plant health and environmental (including climatic) conditions relevant to the use of the product are comparable in the regions concerned.
|
| |
(3) Restrictions on use, necessary in order to avoid exposure of consumers of treated products to risks of dietary contamination in excess of the acceptable daily intake of the residues concerned, and to take account of differences in dietary patterns, shall where appropriate be attached to authorizations granted in accordance with paragraph (2).
|
| |
(4) With the agreement of the applicant, authorizations granted in accordance with paragraph (2) shall be subject to changes in the conditions of use in order to render, in the regions concerned, any non-comparable agricultural, plant health or environmental (including climatic) conditions irrelevant for the purpose of comparability.
|
| |
(5) In granting authorizations in accordance with paragraph (1) or Regulation 15 (2), the competent authority shall attach those conditions and restrictions to each authorization, as are necessary and relevant—
|
| |
( a ) to ensure compliance with Article 4 (1) (b) of the Directive of 1991, and
|
| |
( b ) to ensure the maximum residue levels provisionally established in accordance with paragraph (1) (c) and Regulation 28 are not exceeded.
|
| |
(6) Subject to the provisions of Regulation 36, authorizations granted in accordance with this Regulation shall be for fixed periods of 10 years.
|
|
|
14 Refusal to Recognize Comparability
|
14. (1) Where the competent authority does not accept that claims as to comparability, with respect to the agricultural, plant health and environmental (including climatic) conditions relevant to use of the product in the regions concerned, made in accordance with Regulation 8 (5) (b), are justified, and has required the repetition of one or more tests and analyses, it shall notify the Commission of the grounds on which the repetition of testing or analysis was required.
|
| |
(2) Where the competent authority does not accept that claims as to comparability, with respect to the agricultural, plant health and environmental (including climatic) conditions relevant to use of the product in the regions concerned, made in accordance with Regulation 13 (2), are justified, and has refused to authorize a plant protection product in accordance with that provision, it shall notify the Commission of the grounds on which the authorization was refused.
|
| |
(3) Where a decision different from that of the competent authority is made pursuant to Article 10.3 of the Directive of 1991, the competent authority shall without delay accept the tests and analyses or authorize the placing of the plant protection product on the market, subject in the latter case to any terms which the above decision may set.
|
|
|
15 Authorization for Provisional Periods
|
15. (1) Notwithstanding the provisions of Regulation 13 (1) (a), application may be made in accordance with Regulation 8 for the authorization for a provisional period of the placing on the market and use of a plant protection product containing an active substance not listed in Annex I and not yet available on the market 2 years after the notification of the Directive of 1991.
|
| |
(2) The competent authority may grant such an authorization for a period not exceeding three years, provided that—
|
| |
( a ) following application of Article 6(2) and (3) of the Directive of 1991, it is found that the dossier on the active substance satisfies the requirements of Annexes II and III in relation to the projected uses,
|
| |
( b ) the active substance can satisfy the requirements of Article 5 (1) of the Directive of 1991 and that the plant protection product may be expected to satisfy the requirements of Regulation 13 (1) (b) and (c), and
|
| |
( c ) its packaging and labelling satisfy the provisions of Regulations 23 and 24.
|
| |
(3) The competent authority shall immediately inform the other Member States and the Commission of its assessment of each application considered and of the terms of each authorization granted for a provisional period, giving at least the information provided for in Regulation 22(1).
|
| |
(4) Notwithstanding the provisions of paragraph (2), where on expiry of the three-year period, a decision has not been taken concerning the inclusion of an active substance in Annex I, a further period of provisional authorization may be granted of a duration consistent with the period ordered in accordance with the final paragraph of Article 8 (1) of the Directive of 1991.
|
| |
(5) The granting of authorizations for provisional periods shall be subject to the provisions of Regulation 36.
|
|
|
16 Extension of the Field of Application of Authorized Plant Protection Products
|
16. (1) Official or scientific bodies involved in agricultural activities or professional agricultural organizations and professional users may request that the field of application of a plant protection product already authorised be extended to purposes other than those covered by the authorization.
|
| |
(2) Subject to the provisions of paragraph (4), the competent authority shall grant an extension of the field of application of an authorized plant protection product, when it is in the public interest to do so, where—
|
| |
( a ) an application for an extension of the field of application of an authorized plant protection product, is made and is in the form set out in Part 1 of the Fifth Schedule,
|
| |
( b ) documentation and information, as specified in Part 2 and Part 3 of the Fifth Schedule, to support an extension of the field of application has been submitted by the applicant,
|
| |
( c ) the competent authority has established that the conditions referred to in Article 4(1)(b)(iii), (iv) and (v) of the Directive of 1991, are satisfied, and
|
| |
( d ) the extension of the field of application relates to a use or uses which is or are minor in nature.
|
| |
(3) The competent authority shall ensure that users are fully and specifically informed as to instructions for use, by means of an addition to the labelling or, failing that, by means of an official publication.
|
| |
(4) The granting of an extension of the field of application of an authorized plant protection product shall be subject to the provisions of Regulation 36. Each such extension granted shall expire on the same date as the authorization to which it relates.
|
| |
(5) The provisions of this Regulation shall also apply to plant protection products which are pesticides referred to in Regulation 3 (1) (a) of the Regulations of 1994, which have not yet been authorized in accordance with these Regulations and which are placed on the market in accordance with the Regulations of 1994.
|
|
|
17 Information on Potentially Harmful Effects
|
17. (1) Those to whom an authorization has been granted and those to whom an extension of the field of application has been granted in accordance with Regulation 16, shall immediately notify the competent authority of all new information on the potentially dangerous effects of any plant protection product, or of residues of an active substance, on human or animal health or on ground water, or their potentially dangerous effects on the environment.
|
| |
(2) The competent authority shall require the parties concerned to immediately notify the information provided in accordance with paragraph (1) to the other Member States and to the Commission.
|
|
|
18 Authorization of Plant Protection Products Containing Active Substances on the Market Prior to 25 July 1993, Pending a Decision concerning their inclusion in Annex I
|
18. (1) Subject to the provisions of paragraph (4) and Regulation 19 (7), during the period to 24 July 2003, applications may be made to the competent authority for authorization of plant protection products in accordance with Regulation 8 for products containing active substances not listed in Annex I that are already on the market 2 years after the notification of the Directive of 1991.
|
| |
(2) Pending the review of such active substances in accordance with Article 8 (2) of the Directive of 1991, such applications shall be examined by the competent authority, which shall decide thereon within a reasonable period, provided that it has the necessary scientific and technical resources at its disposal.
|
| |
(3) In deciding on applications made in accordance with paragraph (1), the competent authority shall not authorize the placing on the market and use of any plant protection product unless—
|
| |
( a ) following application of the uniform principles as contained in Annex VI, the requirements of Article 4 (1) (b), (c), (d) and (e) of the Directive of 1991 are satisfied,
|
| |
( b ) maximum residue levels in the agricultural products referred to in the authorization have been provisionally established, and notified to the Commission in accordance with Regulation 22, such maximum residue levels, when adopted pursuant to 28 (1), to remain in force until replaced in accordance with the procedures specified in Article 4 (1) (f) of the Directive of 1991, and
|
| |
( c ) its packaging and labelling satisfy the provisions of Regulations 23 and 24.
|
| |
(4) Subject to the provisions of Regulation 36, an authorization granted in accordance with this Regulation shall be for a fixed period of 10 years.
|
|
|
19 Renewal, Review, Amendment and Cancellation of Authorizations and Extensions of the field of Application of Authorized Plant Protection Products
|
19. (1) Subject to the provisions of paragraph (12), where an application is made for the renewal of an authorization in the form set out in Part 1 of the Sixth Schedule, and is supported with the documentation specified in Regulation 8 (3) and where relevant, the documentation specified in Regulation 8 (5) (b), the authorization shall be renewed where the competent authority has verified that the requirements of Regulation 13 (1) are still satisfied. Renewal shall be granted for the period necessary for such verification, where an application for such renewal has been made.
|
| |
(2) Subject to the provisions of paragraph (12), where an application is made for the renewal of an extension of the field of application of an authorization in the form set out in Part 2 of the Sixth Schedule, and is supported with the documentation specified in Regulation 16 (2) (b), an extension shall be renewed where the competent authority has verified that the requirements of Regulation 16 (2) are still satisfied. Renewal shall be granted for the period necessary for such verification, where an application for such renewal has been made.
|
| |
(3) An authorization may be reviewed at any time specified by the competent authority, if there are indications that any of the requirements provided for in Regulation 13(1) are no longer being satisfied. In such a case the competent authority shall require the holder of the authorization concerned, or party to whom an extension of the field of application was granted in accordance with Regulation 16, to submit further information necessary for the review at a time to be specified by the competent authority. The authorization shall, where necessary, be extended for the periods necessary to provide such further information and to complete the review.
|
| |
(4) Where an application is made by the holder of an authorization for its modification in the form set out in Part 3 of the Sixth Schedule and is supported with a statement as to the reasons therefor and with the documentation specified in Regulation 8 (3), subject to the provisions of paragraph (12), an amendment to the authorization shall be granted where the competent authority has verified that the requirements of Regulation 13(1), Regulation 15 (2) and Regulation 18 (2), as appropriate, are still satisfied.
|
| |
(5) Where an application is made by the holder of an authorization for its cancellation in the form set out in Part 4 of the Sixth Schedule and is supported with a statement as to the reasons therefor, the competent authority shall cancel the authorization.
|
| |
(6) Without prejudice to decisions taken pursuant to Regulation 13 (2), authorizations shall be:
|
| |
( a ) cancelled if it is established that—
|
| |
(i) the requirements for obtaining the authorization are not, or are no longer, satisfied, or
|
| |
(ii) false or misleading particulars were supplied concerning the facts on the basis of which the authorization was granted; or
|
| |
( b ) modified if it is established that on the basis of developments in scientific and technical knowledge the manner of use and amounts used can be modified.
|
| |
(7) Following the evaluation of a dossier as provided for in Article 6 (3) of the Directive of 1991 the competent authority, within 6 months of the completion of the evaluation concerned, shall—
|
| |
( a ) where it has been decided that the active substance does not satisfy the requirements specified in Article 5(1) of the Directive of 1991, cancel any authorization granted in accordance with Regulations 13 or 15 (2) for plant protection products containing the active substance, and
|
| |
( b ) where it has been decided to include the active substance in Annex I of the Directive of 1991, modify any authorization granted in accordance with Regulations 13 and 15 (2) and modify any extension of the field of application of any authorization granted in accordance with Regulation 16, for each plant protection product containing the active substance, such that the conditions and restrictions associated with inclusion of the active substance in Annex I are complied with.
|
| |
(8) Following the review of an active substance in accordance with Article 8 (2) of the Directive of 1991, the competent authority shall—
|
| |
( a ) within 6 months of the completion of each such review, where a decision is taken not to include an active substance in Annex I,
|
| |
(i) in the case of a pesticide referred to in Regulation 3 (1) (a) of the Regulations of 1994, which is a plant protection product and which contains the active substance, cancel each notification, clearance and permission to market granted in accordance with the Regulations of 1994, for the each product concerned, and
|
| |
(ii) cancel each authorization granted in accordance with Regulation 18 and each permission granted in accordance with Regulation 5 (3), for each plant protection product containing the substance;
|
| |
( b ) within 6 months of the completion of each such review, where a decision is taken to include an active substance in Annex I,
|
| |
(i) in the case of a pesticide referred to in Regulation 3 (1) (a) of the Regulations of 1994, which is a plant protection product and which contains the active substance,
|
| |
— cancel each relevant notification, clearance and permission to market granted in accordance with those Regulations, for each product concerned, where in accordance with Regulation 10, information referred to in Regulation 8 (3), may not be used to support its authorization, unless the information concerned is provided in accordance with subparagraph (c),
|
| |
— modify each notification, clearance and permission to market granted in accordance with those Regulations, for each product concerned, such that the conditions and restrictions associated with inclusion of the active substance in Annex I are complied with,
|
| |
(ii) in the case of an authorization granted in accordance with Regulation 18 and a permission granted in accordance with Regulation 5 (3), for a plant protection product containing the active substance,
|
| |
— cancel the authorization or permission to market, as appropriate, where in accordance with Regulation 10, information referred to in Regulation 8 (3), may not be used to support the authorization or permission, unless the information concerned is provided in accordance with subparagraph (c),
|
| |
— modify the authorization or permission to market for each product concerned, such that the conditions and restrictions associated with inclusion of the active substance in Annex I are complied with; and
|
| |
( c ) where a decision is taken to include an active substance in Annex I,
|
| |
(i) require persons marketing a pesticide referred to in Regulation 3 (1) of the Regulations of 1994, which is a plant protection product and which contains the active substance, to make application for its authorization in accordance with these Regulations, at a time specified by the competent authority,
|
| |
(ii) in the case of a permission granted in accordance with Regulation 5 (3), for a plant protection product containing the active substance, require persons marketing the product concerned to make application for its authorization in accordance with these Regulations, at a time specified by the competent authority,
|
| |
(iii) in cases referred to in subparagraphs (i) and (ii), subject to the provisions of paragraph (12), examine, and authorize or not each plant protection product concerned in accordance with these Regulations, within a period of 2 years following the receipt of a complete application,
|
| |
(iv) in cases referred to in subparagraphs (i) and (ii), where complete applications are not received by the dates specified, refuse authorization for each plant protection product concerned, unless
|
| |
— it is established that the necessary supporting documentation is being generated, or
|
| |
— access to such documentation is being negotiated.
|
| |
(9) Where the competent authority has valid reasons to consider that a plant protection product which it has authorized or is bound to authorize in accordance with Regulation 13(1) or (2) or Regulation 15 (2), constitutes a risk to human or animal health or the environment, it shall provisionally restrict or prohibit the use and/or sale of that product. In such instances, the competent authority shall immediately inform the Commission and the other Member States of such action and provide reasons for its decision.
|
| |
(10) Where an authorization is cancelled in accordance with this Regulation, the competent authority shall immediately inform the holder of the authorization of the cancellation. Without prejudice to decisions taken and periods provided for in any decision under Council Directive 79/117/EEC of 21 December 1978 prohibiting the placing on the market and use of plant protection products containing certain active substances 13, as last amended by Directive 90/533/EEC14, or the consequences of decisions taken pursuant to Article 6(1) or Article 8(1) or (2) of the Directive of 1991, notwithstanding the provisions of this Regulation, or the provisions of Regulations 4, 6 and 31, the competent authority may grant a period of grace for the disposal, storage, placing on the market and use of existing stocks, of a length in accordance with the reason for the withdrawal.
|
| |
(11) In the case of a plant protection product for which some or all uses are cancelled or modified pursuant to the provisions of paragraph (8), notwithstanding the provisions of this Regulation, or of Regulations 4, 6 and 31, an authorised officer acting on behalf of the Minister, may, by a notice in writing given to the owner or person in apparent charge or control, permit the controlled placing on the market or use of existing stocks of the plant protection product, subject to such conditions as he may specify in order to minimize any unacceptable risk to man, animals or the environment that might arise from such use.
|
| |
(12) The consideration of applications for the renewal of an authorization in accordance with paragraph (1), for the renewal of an extension of the field of application of an authorization in accordance with paragraph (2), for the modification of an authorization in accordance with paragraph (4) and for authorization in accordance with paragraph (8) (c), shall be subject to the provisions of Regulation 36.
|
| |
13 O.J. No. L33/36 8/2/1979
|
| |
14 O.J. No. L296/63 27/10/1990
|
| |
(13) Authorizations granted in accordance with these Regulations shall be cancelled where there has been a failure to comply with the provisions of Regulation 36 (4).
|
|
|
20 Emergency Authorizations
|
20. (1) Notwithstanding the provisions of Regulations 13, 15 and 18, the competent authority may authorize for a period not exceeding 120 days the placing on the market of plant protection products not complying with the provisions of Regulations 13, 15 or 18 for limited and controlled use if such a measure appears necessary because of an unforeseeable danger which cannot be contained by other means.
|
| |
(2) Where authorizations are granted in accordance with paragraph (1), the competent authority shall immediately inform the other Member States and the Commission of its action.
|
|
|
21 Files on Applications
|
21. (1) The competent authority shall ensure that a file is compiled on each application for authorization of a plant protection product in accordance with Regulations 13, 15 and 18, and on each application for extension in the field of application of an authorized plant protection product in accordance with Regulation 16.
|
| |
(2) Each file shall contain at least—
|
| |
( a ) a copy of the application,
|
| |
( b ) a record of the administrative decisions taken by the competent authority concerning the application,
|
| |
( c ) a record of the administrative decisions taken by the competent authority concerning the particulars and documentation provided for in accordance with Regulation 8 (3), and
|
| |
( d ) a summary of the particulars and documentation provided for in accordance with Regulation 8 (3), (5) (b) and (6).
|
| |
(3) The competent authority shall on request make available to the competent authorities of other Member States and to the Commission the files provided for in paragraph (1), and shall supply to them on request all information necessary for full comprehension of applications, and shall, where requested, ensure that applicants provide a copy of the technical documentation specified in Regulation 8 (3) (a).
|
|
|
22 Information Exchange
|
22. (1) Within a period of one month at the end of each quarter, the competent authority shall inform the competent authorities of the other Member States and the Commission in writing of any plant protection product authorized or for which an authorization has been cancelled, in accordance with the provisions of these Regulations, indicating at least—
|
| |
( a ) the name or business name of the holder of the authorization,
|
| |
( b ) the trade name of the plant protection product,
|
| |
( c ) the type of preparation,
|
| |
( d ) the name and amount of each active substance which it contains,
|
| |
( e ) the use or uses for which it is or was intended,
|
| |
( f ) the maximum residue levels provisionally established where they have not already been set by Community rules,
|
| |
( g ) where relevant, the reasons for cancellation of an authorization, and
|
| |
( h ) the dossier needed for the evaluation of the maximum residue levels provisionally established.
|
| |
(2) The competent authority shall draw up an annual list of the plant protection products authorized in its territory and shall communicate that list to the competent authorities of the other Member States and to the Commission.
|
|
|
23 Packaging
|
23. (1) The packaging of plant protection products authorized in accordance with these Regulations, together with the fastenings and containers used in such packaging and the materials constituting such packaging or fastenings shall comply with the requirements of Article 5(1) of the Directive of 1978, whether or not they are pesticide preparations which are dangerous for the purposes of the Directive of 1978.
|
| |
(2) Where the packaging of a plant protection product to which these Regulations apply includes an inner liner, except where the liner is used as a seal to protect rodenticide baits, or is a water soluble sachet contained in a sealed foil sachet, it shall not be detachable from the rest of the packaging unless it itself complies with the requirements of paragraph (1).
|
| |
(3) In addition to complying with paragraph (1)—
|
| |
( a ) packages containing a plant protection product to which these Regulations apply shall be closed with a distinctive seal in such a way that when the package is opened for the first time the seal is irreparably damaged, and
|
| |
( b ) containers having a capacity not exceeding three litres shall have childproof fastenings designed in accordance with Annex VII where the plant protection product—
|
| |
(i) is intended for domestic or garden use, and
|
| |
(ii) is a pesticide preparation which is a dangerous pesticide for the purposes of the Directive of 1978.
|
|
|
24 Labelling
|
24. (1) The packaging of plant protection products authorized in accordance with these Regulations, or labels attached to packaging, shall comply with the requirements of paragraph (2) as to labelling.
|
| |
(2) Subject to the provisions of paragraph (3), the following information shall be stated clearly and in an indelible form on all packaging which is packaging mentioned in Regulation 23 (1), or on a label on or attached to such packaging in the Irish language or in the English language or in both the Irish and English languages:
|
| |
( a ) the trade name or designation of the plant protection product;
|
| |
( b ) the name and address of the holder of the authorization and the authorization number of the plant protection product and, if different, the name and address of the person responsible for the final packaging and labelling or for the final labelling of the plant protection product on the market;
|
| |
( c ) the name of each active substance as given in the list contained in Annex I to the Directive of 1967, if not included therein, its ISO common name. If the latter is not available, the active substance shall be designated by its chemical designation according to IUPAC rules;
|
| |
( d ) the amount of each active substance so contained expressed—
|
| |
(i) for pesticides which are solids, aerosols, volatile liquids (maximum boiling point 50°C) or viscous liquids (lower limit 1 Pas at 20°C), as a percentage by weight,
|
| |
(ii) for other liquids, as a percentage by weight and in grams per litre at 20°C,
|
| |
(iii) for gases, as a percentage by volume, and
|
| |
(iv) for acids, their amides, esters and salts, on an acid equivalent basis;
|
| |
( e ) the net quantity of plant protection product given in legal units of measurements;
|
| |
( f ) the formulation batch number or some means of identifying it;
|
| |
( g ) the particulars required under Article 6 of the Directive of 1978—
|
| |
(i) the name as given in the list contained in Annex I to the Directive of 1967, of each very toxic, toxic, harmful or corrosive substance, excluding active substances, contained in the plant protection product, in concentrations of more than 0.2 percent in the case of very toxic and toxic substances, 5 percent in the case of harmful substances and 5 percent in the case of corrosive substances,
|
| |
(ii) the symbols and indications as specified in Annex VIII,
|
| |
(iii) phrases indicating the nature of the special risks referred to in Article 6 (2)(h) of the Directive of 1978, selected from among those contained in Annex IX and specified by the competent authority,
|
| |
(iv) phrases providing safety advice concerning the use of the plant protection product, selected by the competent authority from those contained in Annexes X and XI, and
|
| |
(v) for plant protection products classified in accordance with Regulation 5 of the Regulations of 1994, as being very toxic, toxic or harmful, a statement that the packaging must not be re-used for any purpose, except in the case of containers which are specifically designed for re-use, recharging or refilling by the manufacturer or distributor;
|
| |
( h ) first-aid information for use in the event of accidental exposure or ingestion;
|
| |
( i ) the nature of any special risks for humans, animals or the environment, by means of standard phrases selected as appropriate from those given in Annex IV;
|
| |
( j ) safety precautions for the protection of humans, animals or the environment, in the form of standard phrases selected as appropriate from those given in Annex V;
|
| |
( k ) in accordance with Article 16 (5) of the Directive of 1991, additional phrases deemed necessary for the protection of human beings, animals or the environment, where appropriate selected from those set out in Annex XII;
|
| |
( l ) the type of action of the plant protection product (e.g. insecticide, growth regulator, herbicide, etc);
|
| |
( m ) the type of preparation (e.g. wettable powder, emulsifiable concentrate, etc);
|
| |
( n ) the uses for which the plant protection product has been authorized and any specific agricultural, plant health and environmental conditions under which the product may be used or should not be used;
|
| |
( o ) directions for use and the dose rate, expressed in metric units, for each use provided for under the terms of the authorization;
|
| |
( p ) where necessary, the safety interval for each use between application and—
|
| |
(i) sowing or planting of the crop to be protected,
|
| |
(ii) sowing or planting of succeeding crops,
|
| |
(iii) access by humans or animals,
|
| |
(iv) harvesting,
|
| |
(v) use or consumption;
|
| |
( q ) particulars of possible phytotoxicity, varietal susceptibility, and any other direct or indirect adverse side effects on plants or products of plant origin together with the intervals to be observed between application and sowing or planting of—
|
| |
(i) the crop in question, or
|
| |
(ii) subsequent crops;
|
| |
( r ) if accompanied by a leaflet, as provided for in paragraph 3, the sentence Read accompanying instructions before use;
|
| |
( s ) directions for safe disposal of the plant protection product and of the packaging;
|
| |
( t ) the expiry date relevant to normal conditions of storage where the shelf life of the product is limited to less than two years;
|
| |
( u ) where relevant, the category or categories of users to which supply and use is restricted; and
|
| |
( v ) the authorization number, or permission to market number, as appropriate, of the plant protection product.
|
| |
(3) Notwithstanding the provisions of paragraph (2), the information specified in subparagraphs (o), (p) and (q) of paragraph (2) may be included on a separate leaflet accompanying the package if the space available on the package is too small. Such a leaflet shall be regarded as part of the label for the purposes of these Regulations.
|
| |
(4) Labels and packaging of plant protection products shall not bear indications such as non-toxic, harmless, or other similar indication. However, information to the effect that the plant protection product may be used when bees or other non-target species are active, or when crops or weeds are in flower or other such phrases to protect bees or other non-target species may be given on the label, if the authorization relates explicitly to use during the season for bees or other specified organisms and presents minimal hazard to them.
|
| |
(5) The competent authority shall notify the competent authorities of the other Member States and the Commission of each additional phrase specified and the reasons for its specification.
|
|
|
25 Authorization for Trials Purposes
|
25. (1) The placing on the market for use in experiments and tests, of a plant protection product to which these Regulations apply, and use of such a product in experiments and tests, other than in accordance with the provisions of Regulation 26, where—
|
| |
( a ) the testing or experimentation is for research or development purposes, and
|
| |
( b ) the tests or experiments concerned involve any release into the environment of an unauthorized plant protection product, or of an authorized plant protection product for an unauthorized use,
|
| |
are hereby prohibited unless the requirements of this Regulation, in relation to authorization for trial purposes, are complied with, and the experiments and tests are carried out in compliance with the requirements specified in the Seventh Schedule, or in compliance with Irish/European Standard IS/EN 45001.
|
| |
(2) The provisions of this Regulation, in relation to authorization of plant protection products for trials purposes, shall not apply to experiments or tests covered by Part B of Council Directive No. 90/220/EEC of 23 April 1990.15.
|
| |
(3) Every application for authorization of a plant protection product for trials purposes, shall be made by or on behalf of the person responsible for, or on whose behalf, the research and development is to be conducted, subject to the person concerned having an office in a Member State, and shall be in the form set out in Part 1 of the Eight Schedule.
|
| |
(4) Applications for authorization of a plant protection product for trials purposes, which shall be supported with the documentation specified in paragraph (5), shall be in the English language.
|
| |
(5) Every application for authorization of a plant protection product for trials purposes, shall be submitted to the competent authority at least 45 days before the date on which it is intended that the experiments or tests commence, and shall be supported with a dossier satisfying, in the light of current scientific and technical knowledge, the requirements set out in the Ninth Schedule.
|
| |
(6) The tests and analyses conducted for the purposes of compiling dossiers referred to in paragraph (5), shall be carried out under agricultural, plant health and environmental conditions relevant to use of the plant protection product in question and representative of those prevailing where the product is intended to be used, and to the extent that they are carried out in the territory of the state, shall be officially recognized tests and analyses.
|
| |
(7) Notwithstanding paragraph (5), and subject to Regulation 10, applicants shall be exempted from supplying the information relevant to the active substance, except for that identifying the active substance, if—
|
| |
15 O.J. No. L117/15 8/5/1990
|
| |
( a ) it is already listed in Annex I, taking into account the conditions of inclusion in Annex 1, and does not differ significantly in degree of purity and nature of impurities, from the composition registered in the relevant Annex II dossier supporting the inclusion of the active substance in Annex I,
|
| |
( b ) a plant protection product containing the active substance is authorized for a provisional period in accordance with Regulation 15,
|
| |
( c ) a plant protection product containing the active substance is authorized in accordance with Regulation 18, or
|
| |
( d ) a plant protection product containing the active substance has been cleared in accordance with the Regulations of 1994.
|
| |
(8) The competent authority shall examine every application received for the authorization of a plant protection product for trials purposes, and shall decide there on within a reasonable period, provided that it has the necessary scientific and technical resources at its disposal.
|
| |
(9) The competent authority shall not authorize the placing on the market and use of any plant protection product for trials purposes unless it is satisfied that when used in accordance with the conditions and restrictions specified pursuant to paragraphs (10) and (11), it has no harmful effect on human or animal health and no unacceptable influence on the environment.
|
| |
(10) Restrictions on use, necessary in order to avoid harmful effects on human or animal health that may arise—
|
| |
( a ) through exposure of consumers of treated products to risks of dietary contamination—
|
| |
(i) in excess of the Acceptable Daily Intake level (ADI) of the residues concerned, or
|
| |
(ii) due to residues for which the health risks associated with exposure have yet to be established, and
|
| |
( b ) through direct exposure of workers and bystanders—
|
| |
(i) in excess of the Acceptable Operator Exposure Level (AOEL) for the active substance or substances concerned, or
|
| |
(ii) due to exposure to active substances for which the health risks associated with exposure have yet to be established,
|
| |
shall, where appropriate, be attached to authorizations granted in accordance with paragraph (8).
|
| |
(11) In granting authorizations for trials purposes in accordance with paragraph (8), the competent authority shall attach any further conditions and restrictions to each such authorization, as are necessary and relevant to avoid any harmful effect on human or animal health and any unacceptable influence on the environment, to include—
|
| |
( a ) particular packaging and labelling requirements,
|
| |
( b ) restrictions as to the quantity that may be placed on the market and used for trials purposes,
|
| |
( c ) restrictions as to the area or areas that may be treated, and
|
| |
( d ) conditions necessary to ensure that the use for trials purposes is controlled and subject to official supervision.
|
| |
(12) Subject to the provisions of Regulation 36, authorizations for trials purposes—
|
| |
( a ) shall be for fixed periods of 12 months,
|
| |
( b ) shall be varied as to any conditions and restrictions attached, where application for such variation is made in the form set out in Part 2 of the Eight Schedule, and the competent authority is satisfied that the provisions of paragraph (9) will be complied with under the changed conditions or restrictions, and
|
| |
( c ) may be renewed for further fixed periods of 12 months, where application for such renewal is made in the form set out in Part 3 of the Eight Schedule.
|
| |
(13) Experiments and tests conducted in accordance with the conditions and restrictions specified in an authorization for trials purposes, are hereby deemed to have been conducted by officially recognized testing facilities or organizations, for the purposes of these Regulations.
|
|
|
26 Trials Permits
|
26. (1) Subject to paragraph (2), and notwithstanding the provisions of Regulation 25, a "trials permit" shall be granted by the competent authority to a person, organization or official body involved in research or development, for specified premises and sites, to conduct tests and experiments using plant protection products for which for authorization for trials purposes has not been granted, or using authorized plant protection products for a use not yet authorised, where—
|
| |
( a ) application is made by the person, organization or official body, in the form set out in Part 1 of the Tenth Schedule, and
|
| |
( b ) the competent authority is satisfied that the requirements specified in paragraph (3) are satisfied.
|
| |
(2) A person, organization or official body, that holds a trials permit is hereby exempted from the provisions of Regulation 25, in relation to tests and experiments which comply with the requirements of paragraph (6), where they are conducted in accordance with the conditions and restrictions of the trials permit specified pursuant to paragraphs (4) and (5).
|
| |
(3) A trials permit shall not be granted for particular premises or sites, unless the applicant—
|
| |
( a ) owns, or has exclusive control of premises or sites suitable for conducting trials and experiments,
|
| |
( b ) owns, or has available, equipment and other facilities, necessary for conducting trials and experiments, at each such premises or site, and
|
| |
( c ) holds, or an employee of his holds, appropriate professional qualifications.
|
| |
(4) Each trials permit granted shall be subject to the condition that—
|
| |
( a ) tests and experiments conducted in accordance with the trials permit shall be carried out in compliance with the requirements specified in the Seventh Schedule,
|
| |
( b ) unless the tests and experiments are conducted by an organization or laboratory accredited in accordance with Irish/European Standards IS/EN 45002 and 45003 by the Irish Laboratory Accreditation Board to carry out such tests and experiments in accordance with Irish/European Standard IS/EN 45001.
|
| |
(5) Each trials permit granted shall be subject to conditions and restrictions such that the use of plant protection products in tests and experiments conducted in accordance with the trials permit has no harmful effect on human or animal health and no unacceptable influence on the environment. The conditions and restrictions specified for each trials permit shall—
|
| |
( a ) restrict its validity to the premises, site or sites specified in the trials permit,
|
| |
( b ) restrict its validity to tests and experiments conducted under the direct supervision of the professionally qualified personnel specified in the trials permit, and
|
| |
( c ) be conditional on an authorization for trials purposes being obtained, in accordance with Regulation 25, in each instance in which trials or experiments other than those conforming to the requirements of paragraph (6) are to be conducted.
|
| |
(6) A trials permits shall not be valid for tests and experiments involving plant protection products unless—
|
| |
( a ) the conditions and manner of use are encompassed by an existing authorization for a plant protection product containing the same active substance or substances,
|
| |
( b ) use is restricted to crops other than food or feed crops,
|
| |
( c ) the nature of the use or of the active substance is such that residues at harvest are precluded, or
|
| |
( d ) food, feed and forage crops are destroyed by burning or burying to preclude consumption by humans or animals.
|
| |
(7) Subject to the provisions of Regulation 36, a trials permit granted—
|
| |
( a ) shall be for fixed periods of 12 months,
|
| |
( b ) may be varied as to any conditions and restrictions attached, where application for such variation is made in the form set out in Part 2 of the Tenth Schedule, and the competent authority is satisfied that the provisions of paragraph (3) will be complied with under the changed conditions or restrictions, and
|
| |
( c ) may be renewed for a further fixed periods of 12 months, where application for such renewal is made in the form set out in Part 3 of the Tenth Schedule.
|
| |
(8) Experiments and tests conducted in accordance with the conditions and restrictions associated with a trials permit, are hereby deemed to have been conducted by officially recognized testing facilities or organizations, for the purposes of these Regulations.
|
|
|
27 Notification of Imports and Exports
|
27. (1) Unless exempted in accordance with the provisions of paragraph (2), where a person proposes to import into the State a plant protection product to which these Regulations apply, three days notice of the intended importation shall be given in writing, in the form set out in Part 1 of the Eleventh Schedule, to the competent authority on behalf of the importer specifying—
|
| |
( a ) the brand name of the plant protection product,
|
| |
( b ) the port, airport or other place at which it is expected that the plant protection product shall be landed or otherwise brought into the State,
|
| |
( c ) the date on which the plant protection product is expected to be so transferred into the State,
|
| |
( d ) the number of packs which the relevant consignment comprises (if it comprises more than one),
|
| |
( e ) the pack size (given by reference to volume or weight) of the consignment or, in case the consignment comprises more than one pack size, the pack size (so given) of each such pack, and
|
| |
( f ) in the case of a plant protection product being imported into the State, the destination to which the pesticide is consigned or, in lieu thereof, an address at which the plant protection product may be examined, sampled, tested or inspected pursuant to Regulation 30 (1).
|
| |
(2) Where satisfied that plant protection products imported and intended for use in the territory of the State, in the first instance following importation, will be transferred to nominated warehouse or storage facilities, the competent authority may grant an exemption from the requirements of paragraph (1) to particular importers, where
|
| |
( a ) the importer has provided the address of each premises at which plant protection products concerned will be stored following importation, prior to supply or sale,
|
| |
( b ) the importer notifies on an annual basis, details of all imports to the competent authority, in the form set out in Part 2 of the Eleventh Schedule, such notification to be provided by 31 January each year in relation to imports during the previous year.
|
| |
(3) Where a plant protection product to which these Regulations apply, is exported out of the State, the exporter shall notify on an annual basis details of the export to the competent authority, in the form set out in Part 3 of the Eleventh Schedule, such notification to be provided by 31 January each year in relation to exports during the previous year.
|
|
|
28 Provisional Maximum Residue Levels
|
28. (1) The Minister may from time to time specify the maximum levels of residues of plant protection products which may be contained in specified controlled products.
|
| |
(2) The maximum levels of residues of plant protection products specified in accordance with paragraph (1) shall be those established by the competent authority pursuant to subparagraph (1) (c) of Regulation 13, subparagraph (2) (b) of Regulation 15 or subparagraph (2) (b) of Regulation 18 and shall remain in force until—
|
| |
( a ) replaced by maximum levels subsequently specified to give effect to provisional maximum levels established by the Community in accordance with Article 4 (1) ( f ) of the Directive of 1991, or
|
| |
( b ) replaced by maximum levels established pursuant to Directive 76/895/EEC16, Directive 86/362/EEC17, Directive 86/363/EEC18, Directive 90/642/EEC19, Directive or 91/132/EEC20, amending Directive 74/63/EEC21.
|
|
|
29 ..
|
29. (1) A person shall not place on the market any controlled product if—
|
| |
( a ) the product contains within it or on it a residue of a plant protection product, and
|
| |
( b ) the level of such residue exceeds the maximum specified in relation to the controlled product in accordance with Regulation 28(1).
|
| |
(2) A person who contravenes the provisions of paragraph (1) shall be guilty of an offence and shall be liable on summary conviction to a fine not exceeding £1,000 or imprisonment for a term not exceeding 6 months or to both.
|
|
|
30 Inspections, Sampling, Tests and Examinations
|
30. (1) Subject to paragraph (5), an authorised officer may at any reasonable time enter—
|
| |
( a ) any place or premises in which he has reasonable grounds for believing that—
|
| |
16O.J. No. L340/26, 9/12/1976
|
| |
17O.J. No. L221/36, 7/8/1986
|
| |
18O.J. No. L221/43, 7/8/1986
|
| |
19O.J. No. L350/71, 14/12/1990
|
| |
20O.J. No. L66/16, 13/3/1991
|
| |
21O.J. No. L38/31, 11/2/1974
|
| |
(i) a plant protection product is being manufactured, placed on the market, stored or used, or
|
| |
(ii) a controlled product is being produced, placed on the market, processed, stored or used,
|
| |
( b ) any railway wagon, vehicle, ship, vessel, aircraft, container or other thing in which he has reasonable grounds for believing that a plant protection product or a controlled product is being either transported, stored or used, or
|
| |
( c ) any premises in which he has reasonable grounds for believing that there are any books, documents or records relating to any business whose activities consist of or include—
|
| |
(i) the manufacture, placing on the market, storage, transport or use of a plant protection product, or
|
| |
(ii) the production, putting into circulation, processing or storage of any controlled product,
|
| |
and there or at any other place—
|
| |
(iii) make such examinations, tests and inspections, and
|
| |
(iv) take samples in accordance with the methods described in the manual on the development and use of FAO specifications for plant protection products (Food and Agriculture Organization of the United Nations, FAO Plant Production and Protection Paper 85, Fourth Revised Edition), of any plant protection product which he finds in the course of his inspection and which he believes is or may be a plant protection product to which these Regulations apply, and
|
| |
(v) take samples in accordance with Commission Directive 79/700/EEC of 24 July 197922, or the Joint FAO/WHO Food Standards Programme, Codex Alimentarius Commission, recommended method of sampling for the determination of Pesticide Residues (Food and Agriculture Organisation of the United Nations CAC/PR5-1984), where relevant, and in accordance with other internationally accepted procedures in other cases, of any plant, plant product, soil, compost, or take samples from or of any other thing, which he finds in the course of an inspection and which he believes may have been treated or contaminated with a plant protection product to which the Regulations apply,
|
| |
22O.J. No. L207/26 15/8/1979
|
| |
as he may consider appropriate and provided the quantity which a sample taken pursuant to this Regulation comprises is reasonable.
|
| |
(2) A person who has in any place, on any premises or in any railway wagon, vehicle, ship, vessel, aircraft, container or other thing a plant protection product to which these Regulations apply, or a controlled product, shall at all reasonable times—
|
| |
( a ) afford to an authorised officer such facilities and assistance as are reasonably necessary for an inspection and for the taking of samples pursuant to this Regulation,
|
| |
( b ) give an authorised officer any information which he may reasonably require regarding the purchase, importation, storage, transportation, sale, supply or use of any such plant protection product or regarding the production, purchase, importation, processing, transport, storage, sale, supply or use of any controlled product, which is within the person's knowledge or procurement,
|
| |
( c ) produce to an authorised officer any document relating to the raw materials used in the formulation of any plant protection product or relating to the production of any controlled product which the authorised officer may reasonably require and when produced permit the officer to inspect and take extracts from the document.
|
| |
(3) In addition to the foregoing any person who carries on the business of manufacturing, formulating, packaging, processing or marketing a plant protection product for the purposes of the Directive of 1991 shall—
|
| |
( a ) keep records of all transactions regarding the plant protection product,
|
| |
( b ) produce at the request of an authorised officer any records, books or other documents relating to such business which are in his possession or under his control,
|
| |
( c ) permit such an officer to inspect and take extracts from such records, books or other documents and give to the officer any information which is within his knowledge or under his control and which such officer may reasonably require in relation to any entries therein,
|
| |
( d ) afford to any such an officer such facilities and assistance as are reasonably necessary for inspecting the stock of any plant protection product on any premises on which such person carries on such a business,
|
| |
( e ) give to such an officer any information he may reasonably require in relation to such transactions, including, in particular, information which he may reasonably require regarding any plant protection product specified by him.
|
| |
(4) Where a sample is taken pursuant to this Regulation, the authorised officer concerned shall—
|
| |
( a ) divide the sample into 3 parts, each of which he shall seal and mark,
|
| |
( b ) give one part thereof to a designated chemist for analysis in accordance with paragraph (5),
|
| |
( c ) leave with, or send by registered post to, the defendant or his agent, a second part thereof, and
|
| |
( d ) retain the remaining part thereof for possible analysis by the State Chemist in accordance with Regulation 34.
|
| |
(5) Where a designated chemist receives a sample from an authorized officer in pursuance of these Regulations, he shall make analyses thereof in accordance with—
|
| |
( a ) the Joint FAO/WHO Food Standards Programme, Codex Alimentarius Commission, Recommendations for Methods of Analysis of Pesticide Residues (Food and Agriculture Organisation of the United Nations CAC/PR 8-1989), as updated from time to time, or the method included as part of the documentation approved in accordance with Regulation 8, as appropriate, and the Codex Guidelines on Good Practice in Pesticide Residue Analysis (Food and Agriculture Organisation of the United Nations CAC/PR 7-1984), in the case of residues in controlled products, and
|
| |
( b ) the relevant CIPAC method (Collaborative International Pesticides Analytical Council Limited, Handbook Volume I, IA, IB, IC, and D), or the method included as part of the documentation approved in accordance with Regulation 8, as appropriate, in other cases.
|
| |
(6) ( a ) In any proceedings for an offence under these Regulations, the result of any test, examination or analysis of, or any report on, a sample taken pursuant to this Regulation shall not be adduced unless, before the proceedings were instituted, one of the parts into which the sample was divided (as required by paragraph (4)) was left with, or sent by registered post to, the defendant or his agent.
|
| |
( b ) In any proceedings for an offence under these Regulations, evidence of the presence of a plant protection product to which the Regulations apply, in or on equipment capable of use for application of the pesticide, shall be evidence, until the contrary is proved, of the use of the plant protection product by the owner or person in possession of the equipment.
|
| |
( c ) In any proceedings for an offence under these Regulations, evidence of the presence of a residue of a plant protection product to which the Regulations apply, in or on agricultural produce, in soil or compost or in or on surfaces or other materials which may have been treated with or exposed to the plant protection product, shall be evidence, until the contrary is proved, of the use of the plant protection product by the owner, occupier or person in possession, as the case may be.
|
| |
( d ) In any proceedings for an offence under these Regulations, a certificate in the form set out in Part 1 of the Twelfth Schedule showing the results of an analysis shall, until the contrary is shown, be sufficient evidence of the facts certified to therein in relation to—
|
| |
(i) the presence in a plant protection product of any active substance, impurity or formulating ingredient, and the level of any such presence, or
|
| |
(ii) the presence of a residue of a plant protection product and the level of such residues in any controlled product, and
|
| |
a document purporting to be such a certificate shall be deemed, until the contrary is shown, to be such a certificate.
|
| |
( e ) In any proceedings for an offence under these Regulations, each of the documents referred to in subparagraphs (1) (c) (iv) and (v), and in subparagraphs (5) (a) and (b) may be proved by a production of a copy thereof purporting to have been published in the Official Journal of European Communities, by the Food and Agriculture Organization of the United Nations, by the Collaborative International Pesticides Analytical Council Limited, or by the production of the document describing the method, certified by the officer-in-charge of the competent authority as being part of the documentation submitted in accordance with Regulation 8, as appropriate.
|
| |
( f ) For the purpose of these Regulations, the presence of a plant protection product, to which these Regulations apply, on any premises (including any stores) where the business of marketing such a plant protection product is carried on, shall, until the contrary is shown, be sufficient evidence that the plant protection product in question is or was being placed on the market by the owner and by the occupier of such premises.
|
| |
(7) If any person—
|
| |
( a ) tampers with any plant protection product so as to procure that any sample of it taken pursuant to these Regulations does not correctly represent the plant protection product,
|
| |
( b ) tampers with any controlled product so as to procure that any sample of it taken pursuant to these Regulations does not correctly represent the product sampled, or
|
| |
( c ) tampers or interferes with any sample taken pursuant to these Regulations,
|
| |
he shall be guilty of an offence and shall be liable on summary conviction to a fine not exceeding £1,000 or to imprisonment for a term not exceeding 6 months or to both.
|
| |
(8) An authorised officer shall be furnished with a certificate of his appointment as an authorised officer and when exercising any power conferred on him by these Regulations shall, if requested by any person affected, produce the certificate to that person.
|
| |
(9) A designated chemist shall be furnished with a warrant of his appointment by the Minister to carry out analyses as required by these Regulations.
|
|
|
31 Seizure, Retention, Removal and Disposal
|
31. (1) An authorised officer may seize and retain, or seize, remove and retain any plant protection product which he believes is a plant protection product to which these Regulations apply or any controlled product to which these Regulations apply and in relation to which the authorised officer has reasonable grounds for suspecting that there is or has been a failure to comply with any provision of these Regulations.
|
| |
(2) An authorised officer may by a notice in writing given to the owner or to the person in apparent charge or control of a plant protection product or controlled product which has been seized under this Regulation—
|
| |
( a ) require things specified in the notice to be done in relation to the plant protection product or controlled product before it is released by an authorised officer,
|
| |
( b ) in the case of a plant protection product, either—
|
| |
(i) require the disposal of the plant protection product by the person to whom the notice is given, in a manner specified in the notice and at the expense of the owner, or
|
| |
(ii) indicate the authorised officer's intention of disposing of the plant protection product at the expense of the owner,
|
| |
such disposal to be, in either case, such as will prevent the said plant protection product from being placed on the market or used, and
|
| |
( c ) in the case of a controlled product require the disposal of the product by the owner, or person in apparent charge or control of the product, in a manner and within a time specified in the notice and at the expense of the owner, such disposal to be such as will prevent the product being used for human or animal consumption,
|
| |
and in case a notice given under this paragraph requires specified things to be done in relation to a plant protection product or controlled product, the authorised officer shall retain control of the plant protection product or controlled product to which the notice relates until the requirements of the notice have been complied with.
|
| |
(3) Where a notice is given under this Regulation, a person shall not, without the consent of the authorised officer by whom the notice was given sell, move, dispose of or otherwise interfere with the plant protection product or controlled product in any way pending compliance with the requirements of the notice.
|
| |
(4) Any person who is aggrieved by a notice given under paragraph (2), in relation to a plant protection product, which either requires the plant protection product to which it relates to be disposed of or indicates an intention to dispose of such a plant protection product may, not later than the expiration of a period of seven days beginning on the date of the notice, appeal against the notice to the District Court in the District Court District in which the notice has been served.
|
| |
(5) Disposal of a plant protection product pursuant to a notice given under paragraph (2) shall not take place until—
|
| |
( a ) the period during which an appeal under paragraph (4) may be taken against the notice has expired, or
|
| |
( b ) an appeal under that paragraph is determined or withdrawn.
|
| |
(6) (a) Where an appeal is made to the District Court under paragraph (4), that court, if it is satisfied that—
|
| |
(i) the plant protection product to which the relevant notice under this Regulation relates is one to which Regulation 3 applies, and
|
| |
(ii) if the plant protection product was released, it might be placed on the market or used for purposes not authorised in accordance with these Regulations, and
|
| |
(iii) there has been a failure to comply with the provisions of these Regulations—
|
| |
shall order that the plant protection product be disposed of in the manner specified in the notice, or in such other manner as may be specified in the court's order and which, in the opinion of the court, will prevent the plant protection product from being used or placed on the market.
|
| |
( b ) Where an order made by the District Court under this paragraph requires the plant protection product to which it relates to be disposed of by an authorised officer, the cost of such disposal shall be recoverable by the Minister as a simple contract debt in any court of competent jurisdiction from the person who was either the owner or in apparent charge or control of the product at the time of its seizure under this Regulation.
|
| |
(7) Notwithstanding paragraph (2) and the requirements of these Regulations in relation to plant protection products, the method of disposal specified in a notice given under paragraph (2) may include its use subject to such conditions as the authorized officer may specify in the notice, provided that the officer is not aware of any apparent risk to man or the environment by such use.
|
| |
(8) In the case of a notice given under paragraph (2) which indicates an intention to dispose of a plant protection product, the ownership of such a plant protection product shall, in the absence of an appeal by the owner against the notice to the District Court, vest in the Minister on the expiration of a period of 7 days beginning on the date of the notice. In the event of an appeal by the owner against the notice to the District Court, ownership of the plant protection product shall vest in the Minister if the court makes an order under paragraph (5) which requires the plant protection product to be disposed of by an authorised officer.
|
| |
(9) In the case of a notice under paragraph (2) which requires the disposal at the expense of the owner of a plant protection product which has been seized under this Regulation and where there has been a failure to pay, the cost of such disposal shall be recoverable by the Minister as a simple contract debt in any court of competent jurisdiction from the person who was either the owner or in apparent charge or control of the plant protection product at the time of its seizure under this Regulation.
|
| |
(10) Where there has been failure to comply with a requirement of a notice given under paragraph (2) with respect to a controlled product, an authorised officer who in pursuance of this Regulation has seized any controlled product may, on giving notice in writing to the owner, or the person in apparent charge or control of such product of his intention to do so, apply to the District Court in the District Court district in which the notice has been served for an order directing that the controlled product be disposed of (by destruction or otherwise) in a manner, specified in the order, that will prevent its being used for human or animal consumption.
|
| |
(11) Where an application is made under paragraph (10) to the District Court for an order directing the disposal of a controlled product, the Court, if it is satisfied that—
|
| |
(i) the controlled product to which the notice relates contains within it or on it a residue of a plant protection product in excess of the maximum specified in relation to that product in accordance with Regulation 28,
|
| |
(ii) if such product were released, it might be put into circulation contrary to Regulation 29, and
|
| |
(iii) such product if consumed would constitute a danger to human or animal health,
|
| |
shall order that the product be disposed of (by destruction or otherwise) in a manner, specified in the order, that will prevent its being used for human or animal consumption.
|
| |
(12) Where an order is made by the District Court under paragraph (11), the order may provide that the controlled product to which it relates shall be disposed of in the manner specified in the notice given under paragraph (2), or in such other manner as may be specified in the Court's order and which, in the opinion of the Court, will prevent the product being used for human or animal consumption.
|
| |
(13) Where an order made by the District Court under paragraph (11) requires that a product to which it relates be disposed of by an authorised officer, the cost of disposing of the relevant product pursuant to and in accordance with the order shall be recoverable by the Minister as a simple contract debt in any court of competent jurisdiction from the person who was either the owner, or in apparent charge or control of the product, at the time it was seized.
|
|
|
32 General Offenses
|
32. (1) A person who contravenes Regulation 4, 6 or 7 shall be guilty of an offence and shall be liable on summary conviction to a fine not exceeding £1,000, to imprisonment for a term not exceeding 6 months, or to both.
|
| |
(2) A person who —
|
| |
( a ) fails to comply with the requirements of Regulations 7, 8, 9, 17, 23, 24, 25 (1), 26 (4) and (5), 27, 30 (2) and (3), or 31 (3), or
|
| |
( b ) who obstructs or interferes with an authorised officer in the course of exercising a power conferred on him by Regulations 30, 31 or 37, or
|
| |
( c ) who in the context of Regulation 8, 11 (1), 16 (2) (b), 17, 19 (3), 25 (5), 26 (2) (a) or 27, submits false or misleading information, or who gives false information when requested to provide information under Regulation 37,
|
| |
shall be guilty of an offence and shall be liable on summary conviction to a fine not exceeding £1,000, to imprisonment for a term not exceeding six months, or to both.
|
|
|
33 Prosecutions and Specific Rules of Evidence
|
33. (1) An offence under these Regulations may be prosecuted by the Minister.
|
| |
(2) In proceedings for an offence under Regulation 32(1) it shall be a defence for the defendant to prove that the plant protection product to which the offence relates, was either in stock or purchased by him during the twelve month period following—
|
| |
( a ) its authorization in accordance with these Regulations, or
|
| |
( b ) the date of its clearance in accordance with the provisions of the Regulations of 1994, if purchased prior to the date of implementation of these Regulations.
|
|
|
34 Referee Analyses
|
34. (1) Where an appeal is made to the District Court concerning the results of any analysis made under Regulation 30 (5), the third part of the sample shall, if the defendant so requests, be analyzed by the State Chemist.
|
| |
(2) The State Chemist shall in making an analysis comply with the methods of analysis that apply in the particular case as specified in Regulation 30 (5) and issue a certificate in the form set out in Part 2 of the Twelfth Schedule to the defendant and to the designated chemist concerned.
|
|
|
35 ..
|
35. The defendant shall be liable for the cost of the analysis carried out by the State Chemist under Regulation 34 in the event that the results of that analysis confirm that there has been a breach of these Regulations.
|
|
|
36 Fees
|
36. (1) The consideration of every application for the authorization of a plant protection product, for the renewal or modification of an authorization granted, for an extension in the field of use of an authorized plant protection product, for the renewal of an extension granted, for the authorization of a plant protection product for trials purposes, for the renewal or variation in the conditions or restrictions of such an authorization, for a trials permit, for the renewal or variation in the conditions or restrictions of a trials permit, for the inclusion of an active substance in Annex I, or for the modification of the conditions or restrictions associated with any such inclusion, shall be subject to the payment of the following fees at the times specified, that is to say—
|
| |
( a ) in the case of an application for authorization of a plant protection product in accordance with Regulation 13, 15, or 18, for inclusion of an active substance in Annex I in accordance with Article 6 of the Directive, or for a modification of the conditions or restrictions associated with any such inclusion, the fee, or fees as appropriate, set out in column (2) of Part 1 of the Thirteenth Schedule, payable to the Minister in respect of the evaluation of the dossiers set out in column (1) of the said Part 1;
|
| |
( b ) in the case of an application for the renewal of an authorization in accordance with Regulation 19 (1), the fee, or fees as appropriate, set out in column (2) of Part 1 of the Thirteenth Schedule, payable to the Minister in respect of the evaluation of the dossiers set out in column (1) of the said Part 1;
|
| |
( c ) in the case of an application for the modification of an authorization in accordance with Regulation 19 (4), the fee, or fees as appropriate, set out in column (2) of Part 2 of the Thirteenth Schedule, payable to the Minister in respect of the each such modification in relation to a category set out in column (1) of the said Part 2;
|
| |
( d ) an initial fee of £100 payable to the Minister shall accompany each application for the authorization of a plant protection product or for the renewal or modification of an authorisation, which payment shall be offset against the fees mentioned in subparagraphs (a), (b) and (c) (evaluation of each application shall not be undertaken until the balance of the relevant fee is paid);
|
| |
( e ) a fee of £150 shall be payable to the Minister in each case where an application is made for a minor amendment to the packaging and labelling or to the documentation, information and materials submitted in accordance with Regulation 8 (3), where it is considered no hazard evaluation or evaluation of the performance of the plant protection product is involved or required, which fee shall not be offset against the fees mentioned in subparagraphs (a), (b) and (c);
|
| |
( f ) a fee of £500 shall be payable to the Minister in each case where an application is made for a permission to market in accordance with Regulation 5 (3), which fee shall not be offset against the fees mentioned in subparagraphs (a), (b) and (c);
|
| |
( g ) a fee of £200 shall be payable to the Minister in each case where an application is made for an extension in field of use of an authorized plant protection product, in accordance with Regulation 16, which fee shall not be offset against the fees mentioned in subparagraphs (a), (b) and (c); and
|
| |
( h ) a fee of £100 shall be payable to the Minister in each case where an application is made for the renewal of an extension in field of use of an authorized plant protection product, in accordance with Regulation 19 (2), which fee shall not be offset against the fees mentioned in subparagraphs (a), (b) and (c).
|
| |
(2) ( a ) The consideration of every application for the authorization of a plant protection product for trials purposes in accordance with the provisions of Regulation 25 shall be subject to the payment of a fee of £250 payable to the Minister.
|
| |
( b ) The renewal of an authorization of a plant protection product for trials purposes in accordance with the provisions of Regulation 25 shall be subject to the payment of a fee of £50 payable to the Minister.
|
| |
( c ) The variation of the conditions or restrictions of an authorization of a plant protection product for trials purposes in accordance with the provisions of Regulation 25 shall be subject to the payment of a fee of £50 payable to the Minister.
|
| |
(3) ( a ) The consideration of every application for a trials permit in accordance with the provisions of Regulation 26 shall be subject to the payment of a fee of £1,000 payable to the Minister.
|
| |
( b ) The renewal of a trials permit in accordance with the provisions of Regulation 26 shall be subject to the payment of a fee of £250 payable to the Minister.
|
| |
( c ) The variation of the conditions or restrictions of a trials permit in accordance with the provisions of Regulation 26 shall be subject to the payment of a fee of £250 payable to the Minister.
|
| |
(4) Authorization of a plant protection product in accordance with the requirements of these Regulations shall be revoked if, in the case such plant protection product continues to be placed on the market or used, there is a failure to pay the annual renewal fee set out in Part 3 of the Thirteenth Schedule, within 30 days of the fee falling due, but renewal of authorization of a plant protection product may be granted where application is made more than 30 days but not more than 60 days after the renewal fell due on payment to the Minister of the late annual renewal fee set out in Part 3 of the Thirteenth Schedule.
|
| |
(5) In the case of a plant protection product already on the market at the time of the commencement of these Regulations, the Minister may reduce the fee payable under paragraph (1), on a request being made to him in that behalf, where he is satisfied that the wholesale sales of the plant protection product during the previous 12 month period did not exceed 50 times the fee payable to the Minister in accordance with paragraph (1).
|
| |
(6) In the case of a plant protection product which is being placed on the market for the first time, the Minister may refund part of the fee payable in accordance with paragraph (1), on a request being made to him in that behalf, where he is satisfied that the wholesale sales of the plant protection product during the 12 month period following the date of its authorization in accordance with Regulation 13, 15 or 18, did not exceed 50 times the fee payable to the Minister in accordance with paragraph (1).
|
| |
(7) In the case of an application for the authorization of a plant protection product for trials purposes, the fee payable under paragraph (2) may be reduced by the Minister on a request being made to him in that behalf where the potential area of use is limited to a specialised area or specialised areas of use.
|
| |
(8) A fee payable under these Regulations may be recovered by the Minister as a simple contract debt in any court of competent jurisdiction.
|
|
|
37 ..
|
37. A person who makes a claim for a reduction or a refund of fees in accordance with paragraphs Regulation 36 (5), (6) or (7) shall, at all reasonable times—
|
| |
( a ) produce, at the request of an authorised officer, any records, books or other documents which are in his possession or under his control which substantiate such a claim,
|
| |
( b ) permit the officer to inspect and take extracts from such records, books or other documents and give to the officer any information which is within his knowledge or under his control and which such officer may reasonably require for the purpose of verifying the claim,
|
| |
( c ) afford to such an officer such facilities and assistance as are reasonably necessary for inspecting the stock of the relevant plant protection product if the officer considers such inspection is necessary for the purpose of verifying the claim.
|
| |
FIRST SCHEDULE
|
| |
Part 1
|
| |
Annex II
|
| |
(Annex II to the Directive of 1991, as amended by Commission Directive No 93/71/EEC of 27 July 1993)
|
| |
REQUIREMENTS FOR THE DOSSIER TO BE SUBMITTED FOR THE INCLUSION OF AN ACTIVE SUBSTANCE IN ANNEX I
|
| |
Introduction
|
| |
The information required shall:
|
| | |
1.1
|
Include a technical dossier supplying the information necessary for evaluating the foreseeable risks, whether immediate or delayed, which the substance may entail for humans, animals and the environment and containing at least the information and results of the studies referred to below;
|
|
1.2
|
where relevant, be generated using test guidelines referred to or described in this Annex, in the case of studies initiated before the adoption of the modification of this Annex, the information shall be generated using suitable internationally or nationally validated test guidelines or, in the absence thereof, test guidelines accepted by the competent authority;
|
|
1.3
|
in the event of a test guideline being inappropriate or not described, or where another one than those referred to in this annex has been used, include a justification, which is acceptable to the competent authority for the guideline used;
|
|
1.4
|
include, when required by the competent authority, a full description of test guidelines used, except if they are referred to or described in this Annex, and a full description of any deviations from them including a justification, which is acceptable to the competent authority, for these deviations;
|
|
1.5
|
include a full and unbiased report of the studies conducted as well as a full description of them or a justification, which is acceptable to the competent authority where:
|
|
|
— particular data and information which would not be necessary owing to the nature of the product or its proposed uses, are not provided, or
|
|
|
— it is not scientifically necessary, or technically possible to supply information and data;
|
|
1.6
|
where relevant, have been generated in accordance with the requirements of Directive 86/609/EEC23, of 24 November 1986, on the approximation of laws, regulations and administrative provisions of the Member States regarding the protection of animals used for experimental and other scientific purposes.
|
|
| |
|
| |
23 O.J. No. L358/1,18/12/1986
|
| | |
2.1
|
Tests and analyses must be conducted in accordance with the principles laid down in Directive 87/18/EEC24 of 18 December 1986, on the harmonization of laws, regulations and administrative provisions relating to the application of the principles of good laboratory practice and the verification of their application for tests on chemical substances, where testing is done to obtain data on the properties and/or safety with respect of human or animal health or the environment.
|
|
2.2
|
Notwithstanding the provisions of 2.1, during the period to 31 December 1999, tests and analyses done to obtain data on the properties and/or safety with respect to honeybees and beneficial arthropods other than bees may have been conducted by officially recognized testing facilities or organisations, in accordance with the principles laid down in the Sixth Schedule, or in compliance with Irish/European Standard IS/EN 45001, where they are conducted within the territory of the state, and in accordance with the requirements of points 2.2 and 2.3 of the introduction to Annex III to Directive 93/71/EEC, where they are conducted outside the territory of the state.
|
|
| |
PART A
|
| |
Chemical substances
|
| | |
1
|
Identity of the active substance
|
|
|
The information provided must be sufficient to identify with precision each active substance, to define it in terms of its specification and to characterize it as to its nature. The information and data referred to, unless otherwise specified, are required for all active substances.
|
|
1.1
|
Applicant (name, address, etc.)
|
|
|
The name and address of the applicant (permanent community address) must be provided as must the name, position, telephone and telefax number of the appropriate person to contact.
|
|
|
Where, in addition, the applicant has an office, agent or representative in the territory of the State, the name and address of the local office, agent or representative must be provided, as must the name, position, telephone and telefax number of the appropriate person to contact.
|
|
1.2
|
Manufacturer (name, address, including location of plant)
|
|
|
The name and address of the manufacturer or manufacturers of the active substance must be provided as must the name and address of each manufacturing plant in which the active substance is manufactured. A contact point (preferably a central contact point, to include name, telephone and telefax number) must be provided, with a view to providing updating information and responding to queries arising, regarding manufacturing technology, processes and the quality of product (including where relevant, individual batches). Where following inclusion of the active substance in Annex I, there are changes in the location or number of manufacturers, the information required must again be notified to the Commission and the Member States.
|
|
| |
24O.J. No. L15/3, 17/01/1987
|
| | |
1.3
|
Common name proposed or ISO-accepted, and synonyms
|
|
|
The ISO common name, or proposed ISO common name and where relevant, other proposed or accepted common names (synonyms), including the name (title) of the nomenclature authority concerned, must be provided.
|
|
1.4
|
Chemical name (IUPAC and CA) nomenclature
|
|
|
The chemical name as given in Annex I to the Directive of 1967, or, if not included in that Directive, in accordance with both IUPAC and CA nomenclature, must be provided.
|
|
1.5
|
Manufacturer's development code number(s)
|
|
|
Code numbers used to identify the active substance and, where available, formulations containing the active substance, during development work, must be reported. For each code number reported, the material to which it relates, the period for which it was used, and the Member States or other countries in which it was used and is being used, must be stated.
|
|
1.6
|
CAS, EEC and CIPAC numbers (if available)
|
|
|
Chemical Abstracts, EEC (EINECS or ELINCS), and CIPAC numbers, where they exist, must be reported.
|
|
1.7
|
Molecular and structural formula, molecular mass
|
|
|
The molecular formula, molecular mass and structural formula of the active substance, and where relevant, the structural formula of each stereo and optical isomer present in the active substance, must be provided.
|
|
1.8
|
Method of manufacture (synthesis pathway) of the active substances
|
|
|
The method of manufacturer, in terms of the identity of the starting materials, the chemical pathways involved, and the identity of by-products and impurities present in the final product, must be provided, for each manufacturing plant. Generally process engineering information is not required.
|
|
|
Where the information provided relates to a pilot plant production system, the information required must again be provided once industrial scale production methods and procedures have stabilized.
|
|
1.9
|
Specification of purity of the active substance in g/kg
|
|
|
The minimum content in g/kg of pure active substance (excluding inactive isomers) in the manufactured material used for production of formulated products, must be reported.
|
|
|
Where the information provided relates to a pilot plant production system, the information required must again be provided to the Commission and the Member States once industrial scale production methods and procedures have stabilized, if production changes result in a changed specification of purity.
|
|
1.10
|
Identity of isomers, impurities and additives (e.g. stabilizers), together with the structural formula and the content expressed as g/kg
|
|
|
The maximum content in g/kg of inactive isomers as well as the ratio of the content of isomers/diastereoisomers, where relevant, must be provided. In addition, the maximum content in g/kg of each further component other than additives, including by-products, and impurities, must be provided. In the case of additives the content in g/kg must be provided.
|
|
|
For each component, present in quantities of 1 g/kg or more, the following information, where relevant, must be provided—
|
|
|
— chemical name according to IUPAC and CA nomenclature;
|
|
|
— ISO common name or proposed common name if available;
|
|
|
— CAS number, EEC (EINECS or ELINCS) number, and CIPAC number if available;
|
|
|
— molecular and structural formula;
|
|
|
— molecular mass; and
|
|
|
— maximum content in g/kg.
|
|
|
Where the manufacturing process is such that impurities and by-products which are particularly undesirable because of their toxicological, ecotoxicological or environmental properties could be present in the active substance, the content of each such compound must be determined and reported. In such cases, the analytical methods used and the limits of determination, which must be sufficiently low, for each compound of concern, must be reported. Additionally the following information, where relevant, must be provided—
|
|
|
— chemical name according to IUPAC and CA nomenclature;
|
|
|
— ISO common name or proposed common name if available;
|
|
|
— CAS number, EEC (EINECS or ELINCS) number, and CIPAC number if available;
|
|
|
— molecular and structural formula;
|
|
|
— molecular mass; and
|
|
|
— maximum content in g/kg.
|
|
|
Where the information provided relates to a pilot plant production system, the information required must again be provided once industrial scale production methods and procedures have stabilised, if the production changes result in a changed specification of purity.
|
|
|
Where the information provided does not fully identify a component viz. condensates, detailed information on the composition must be provided for each such component. The trade name of components added to the active substance, prior to manufacture of formulated product, to preserve stability and facilitate ease of handling, where they are used, must also be provided. Additionally the following information, where relevant, must be provided for such additives—
|
|
|
— chemical name according to IUPAC and CA nomenclature;
|
|
|
— ISO common name or proposed common name if available;
|
|
|
— CAS number, EEC (EINECS or ELINCS) number, and CIPAC number if available;
|
|
|
— molecular and structural formula;
|
|
|
— molecular mass; and
|
|
|
— maximum content in g/kg.
|
|
|
For added components, other than active substances and other than impurities resulting from the manufacturing process, the function of the component (additive) must be given—
|
|
|
antifoaming agent
|
buffer
|
|
|
antifreeze
|
dispersing agent
|
|
|
binder
|
stabiliser
|
|
|
other (specify)
|
|
|
1.11
|
Analytical profile of batches
|
|
|
Representative samples of the active substance must be analyzed for content of pure active substance, inactive isomers, impurities and additives, as appropriate. The analytical results reported must include quantitative data, in terms of g/kg content, for all components present in quantities of more than 1 g/kg and typically should account for at least 98% of the material analyzed. The actual content of components which are particularly undesirable because of their toxicological, ecotoxicological or environmental properties, must be determined and reported. Data reported must include the results of the analysis of individual samples and a summary of that data, to show the minimum or maximum and typical content of each relevant component, as appropriate.
|
|
|
Where an active substance is produced in different plants this information must be provided for each of the plants separately.
|
|
|
In addition, where available and relevant, samples of the active substance produced in laboratory scale or pilot production systems, must be analyzed, if such material was used in generating toxicological or ecotoxicological data.
|
|
2
|
Physical and chemical properties of the active substance
|
|
|
(i) The information provided, must describe they physical and chemical properties of active substances and together with other relevant information, must serve to characterize them. In particular, the information provided must permit—
|
|
|
— physical, chemical, and technical hazards associated with active substances, to be identified;
|
|
|
— classification of active substance as to hazard;
|
|
|
— appropriate restrictions and conditions to be associated with inclusions in Annex I to be selected; and
|
|
|
— appropriate risk and safety phrases to be specified.
|
|
|
The information and data referred to are required for all active substances, except where otherwise specified.
|
|
|
(ii) The information provided, taken together with that provided for relevant preparations, must permit the physical, chemical and technical hazards associated with preparations, to be identified, permit preparations to be classified, and demonstrate that preparations can be used without unnecessary difficulty, and be such that exposure of man, animals, and the environment is minimized, taking account of manner of use.
|
|
|
(iii) The extent to which active substances for which inclusion in Annex I is sought, comply with relevant FAO specifications, must be stated. Divergences from FAO specifications must be described in detail, and justified.
|
|
|
(iv) In certain specified instances, tests must be conducted using purified active substance of stated specification. In such cases the principles of the method(s) of purification used must be reported. The purity of such test material, which must be as high as can be achieved using the best available technology, must be reported. A reasoned justification must be provided in cases where the degree of purity achieved is less than 980 g/kg. Such justification must demonstrate that all technically feasible and reasonable possibilities for the production of the pure active substance have been exhausted.
|
|
2.1
|
Melting point and boiling point
|
|
2.1.1
|
The melting point or where appropriate the freezing or solidification point of purified active substance must be determined in accordance with EEC Method A 1 and be reported. Measurements should be taken up to 360°C.
|
|
2.1.2
|
Where appropriate, the boiling point of purified active substances must be determined in accordance with EEC Method A 2 and be reported. Measurements should be taken up to 360°C.
|
|
2.1.3
|
Where melting point and/or boiling point cannot be determined because of decomposition or sublimation, the temperature at which decomposition or sublimation occurs, must be reported.
|
|
2.2
|
Relative density
|
|
|
In the case of active substances which are liquids or solids, the relative density of the purified active substance must be determined in accordance with EEC Method A3 and be reported.
|
|
2.3
|
Vapour pressure (In Pa), volatility (e.g. Henry's law constant)
|
|
2.3.1
|
The vapour pressure of purified activie substance must be determined in accordance with EEC Method A4 and be reported. Where vapour pressure is less than 10-5 Pa, the vapour pressure at 20 or 25°C may be estimated using a vapour pressure curve.
|
|
2.3.2
|
In the case of activie substnaces which are solids or liquids, volatility (Henry's law constant) of purified active substance must be determined or calculated from its water solubility and vapour pressure and be reported (in PA x m3 x mol-1).
|
|
2.4
|
Appearances (physical state, colour and odour; if known)
|
|
2.4.1
|
A description of both the colour, if any, and the physical state of both the active substance as manufactured and the purified active substance, must be provided.
|
|
2.4.2
|
A description of any odour associated with the active substance as manufactured and with the purified active substance, noted when handling the materials in laboratories or production plants, must be reported.
|
|
2.5
|
Spectra (UV/VIS, IR, NMR, MS), molecular extinction at relevant wavelengths
|
|
2.5.1
|
The following spectra including a table of signal characteristics needed for interpretation must be determined and reported: Ultraviolet/Visible (UV/VIS), infrared (IR), nuclear magnetic resonance (NMR), and mass spectra (MS) of purified active substance. Molecular extinction at relevant wavelengths, must be determined and reported. The wavelengths at which UV/visible molecular extinction occurs are to be determined and reported and must include, where appropriate, a wavelength at the highest absorption value above 290 nm.
|
|
|
In the case of active substances which are resolved optical isomers their optical purity must be measured and reported.
|
|
2.5.2
|
The UV/visible absorption spectra, IR, NMR and MS spectra, where necessary for the identification of impurities considered to be of toxicological, ecotoxicological or environmental significance, must be determined and reported.
|
|
2.6
|
Solubility in water including effect of pH (4 to 10) on solubility
|
|
|
The water solubility of purified active substances under atmospheric pressure must be determined in accordance with EEC Method A 6 and be reported. These water solubility determinations must be made in the neutral range (i.e. in distilled water in equilibrium with atmospheric carbon dioxide). Where the active substance is capable of forming ions, determinations must also be made in the acidic range (pH 4 to 6) and in the alkaline range (pH 8 to 10), and be reported. Where the stability of the active substances in aqueous media is such that water solubility cannot be determined, a justification based on test data must be provided.
|
|
2.7
|
Solubility in organic solvents
|
|
|
The solubility of active substances, as manufactured, in the following organic solvents at 15 to 25°C must be determined and reported if less than 250 g/kg; the temperature applied must be specified:
|
|
|
Aliphatic hydrocarbon
|
—preferably n-heptane
|
|
|
Aromatic hydrocarbon
|
—preferably xylene
|
|
|
Halogenated hydrocarbon
|
—preferably 1, 2-dichlorethane
|
|
|
Alcohol
|
—preferably methanol or isopropyl acetone
|
|
|
Ketone
|
—preferably acetone
|
|
|
Ester
|
—preferably ethyl acetate
|
|
|
If for a particular active substance, one or more of these solvents is unsuitable (e.g. reacts with test material), alternative solvents can be used instead. In such cases, choices made must be justified in terms of their structure and polarity.
|
|
2.8
|
Partition co-efficient n-octanol/ water including effect of pH (4 to 10)
|
|
|
The n-octanol/water partition coefficient of purified active substance must be determined in accordance with EEC Method A 8 and be reported. The effect of pH (4 to 10) must be investigated when the substance is acidic or basic as defined by its pka value ( 2 for bases).
|
|
2.9
|
Stability in water, hydrolysis rate, photochemical degradation, quantum yield and identity of breakdown product(s), dissociation constant including effect of pH (4 to 9)
|
|
2.9.1
|
The hydrolysis rate of purified active substances (usually radiolabelled active substance,> 95% purity), for each of the pH values 4, 7 and 9, under sterile conditions, in the absence of light, must be determined in accordance with EEC Method C 7 and be reported. For substances with a low rate of hydrolysis, the rate can be determined at 50°C, or another appropriate temperature. If degradation is observed at 50°C, degradation rate at another temperature must be determined, and an Arrhenius plot must be constructed to permit an estimate to be made of hydrolysis products formed and the rate constant observed, must be reported. The estimated DT50 value must also be reported.
|
|
2.9.2
|
For compounds with a molar (decadic) absorption coefficient (ε) > 10 (1 x x mol-1 x xcm-1) at a wavelength λ > 290 nm, direct phototransformation in purified (e.g. distilled) water at 20 to 25°C, of purified active substance usually radio labelled using artificial light under sterile conditions, if necessary using a solubilizer, must be determined and reported. Sensitizers such as acetone must not be used as a co-solvent or solubilizer. The light source must simulate sunlight and be equipped with filters to exclude radiation at wavelengths λ > 10% of the active substance added, a mass balance to account for at least 90% of the applied radioactivity, as well as photochemical halflife must be reported.
|
|
2.9.3
|
Where necessary to investigate direct phototransformation, the quantum yield of direct photodegradation in water must be determined and reported, together with calculations to estimate theoretical lifetime of the active substance in the top layer of aqueous systems and the real lifetime of the substance. The method to be used is described in the Annex to the FAO Revised Guidelines on Environmental Criteria for the Registration of Pesticides.
|
|
2.9.4
|
Where dissociation in water occurs, the dissociation constants(s) (pKa values) of purified active substances must be determined in accordance with OECD Test Guideline 112 and be reported. The identity of the dissociated species formed, based on theoretical considerations, must be reported. If the active substance is a salt, the pKa value of the active principle must be given.
|
|
2.10
|
Stability in air, photochemical degradation, identity of breakdown product(s)
|
|
|
An estimation of the photochemical oxidative degradation (indirect phototransformation) of the active substance(s), must be submitted.
|
|
2.11
|
Flammability including auto-flammability
|
|
2.11.1
|
The flammability of active substances as manufactured, which are solids, gases, or are substances which evolve highly flammable gases, must be determined in accordance with EEC Methods A 10, A 11 or A 12, as appropriate, and be reported.
|
|
2.11.2
|
The auto-flammability of active substances as manufactured must be determined in accordance with EEC Method A 15 or A 16, as appropriate, and/or, where necessary, according to the UN-Bowes-Cameron-Cage-Test (UN-Recommendations on the Transport of Dangerous Goods, Chapter 14, Nr. 14.3.4), and be reported.
|
|
2.12
|
Flash point
|
|
|
The flash point of active substances as manufactured with a melting point below 40°C, must be determined in accordance with EEC Method A 9 and be reported; only closed cup methods should be used.
|
|
2.13
|
Explosive properties
|
|
|
The explosive properties of active substances as manufactured, must be determined in accordance with EEC Method A 14, where appropriate, and be reported.
|
|
2.14
|
Surface tension
|
|
|
The surface tension of active substances must be determined in accordance EEC Method A 5 and be reported.
|
|
2.15
|
Oxidizing properties
|
|
|
The oxidizing properties of active substances as manufactured, must be determined in accordance with EEC Method A 17 and be reported, except where examination of their structural formulae, establish beyond reasonable doubt that the active substance concerned is incapable of reacting exothermically with a combustible material. In such cases, it is sufficient to provide that information as justification for not determining the oxidizing properties of the substance.
|
|
3
|
Further information on the active substance
|
|
|
(i) The information provided must describe the intended purposes for which preparations containing the active substance are used, or are to be used and the dose and manner of their use or proposed use.
|
|
|
(ii) The information provided must specify the normal methods and precautions to be followed, in the handling, storage and transport of the active substance.
|
|
|
(iii) The studies, data and information submitted, together with other relevant studies, data and information, must both specify and justify the methods and precautions to be followed in the event of fire. The possible products of combustion in the event of fire should be estimated, based on the chemical structure and the chemical and physical properties of the active substance.
|
|
|
(iv) The studies, data and information submitted, together with other relevant studies, data and information, must demonstrate the suitability of measures proposed for use in emergency situations.
|
|
|
(v) The information and data referred to are required for all active substances, except where otherwise specified.
|
|
3.1
|
Function, e.g. fungicide, herbicide, insecticide, repellant, growth regulator
|
|
|
The function must be specified from among the following:
|
|
|
acaricide
|
plant growth regulator
|
|
|
bactericide
|
repellant
|
|
|
fungicide
|
rodenticide
|
|
|
herbicide
|
semio-chemicals
|
|
|
insecticide
|
talpicide
|
|
|
molluscicide
|
viricide
|
|
|
nematicide
|
other (must be specified)
|
|
3.2
|
Effects on harmful organisms, e.g. contact poison, inhalation poison, stomach poison, fungitoxic or fungistatic, etc. systemic or not in plants
|
|
3.2.1
|
The nature of the effects on harmful organisms must be stated:
|
|
|
contact action
|
|
|
stomach action
|
|
|
inhalation action
|
|
|
fungitoxic action
|
|
|
fungistatic action
|
|
|
desiccant
|
|
|
reproduction inhibitor
|
|
|
other (must be specified)
|
|
3.2.2
|
It must be stated whether or not the active substance is translocated in plants and where relevant whether such translocation is apoplastic, symplastic or both.
|
|
3.3
|
Field of use envisaged, e.g. field, protected crops, storage of plant products, home gardening
|
|
|
The field(s) of use, existing and proposed, for preparations containing the active substance must be specified from among the following:
|
|
|
Field use
|
— Agriculture
|
|
|
|
— Horticulture
|
|
|
|
— Forestry
|
|
|
|
— Viticulture
|
|
|
Protected crops
|
|
|
Amenity
|
|
|
Weed control on non-cultivated areas
|
|
|
Home gardening
|
|
|
House plants
|
|
|
Plant products storage practice
|
|
|
Other (specify)
|
|
3.4
|
Harmful organisms controlled and crops or products protected or treated
|
|
3.4.1
|
Details of existing and the intended use in terms of crops, groups of crops, plants, or plant products treated and where relevant protected, must be provided.
|
|
3.4.2
|
Where relevant, details of harmful organisms against which protection is afforded, must be provided.
|
|
3.4.3
|
Where relevant, effects achieved e.g. sprout suppression, retardation of ripening, reduction in stem length, enhanced fertilization etc., must be reported.
|
|
3.5
|
Mode of action
|
|
3.5.1
|
To the extent that it has been elucidated, a statement must be provided as to the mode of action of the active substance in terms, where relevant, of the biochemical and physiological mechanism(s) and biochemical pathway(s) involved. Where available, the results of relevant experimental studies must be reported.
|
|
3.5.2
|
Where it is known that to exert its intended effect, the active substance must be converted to a metabolite or degradation product following application or use of preparations containing it, the following information, cross referenced to and drawing on information provided in the context of paragraphs 5.6, 5.11, 6.1, 6.2, 6.8, 7.1, 7.2 and 9, where relevant, must be provided for the active metabolite or degradation product—
|
|
|
— chemical name according to IUPAC and CA nomenclature;
|
|
|
— ISO common name or proposed common name;
|
|
|
— CAS number, EEC (EINECS or ELINCS) number, and CIPAC number if available;
|
|
|
— empirical and structural formula; and
|
|
|
— molecular mass.
|
|
3.5.3
|
Available information relating to the formation of active metabolites and degradation products, must be provided, to include —
|
|
|
— the processes, mechanisms and reactions involved;
|
|
|
— kinetic and other data concerning the rate of conversion and if known the rate limiting step; and
|
|
|
— environmental and other factors effecting the rate and extent of conversion.
|
|
3.6
|
Information on the occurrence or possible occurrence of the development of resistance and appropriate management strategies
|
|
|
Where available information on possible occurrence of the development of resistance or cross-resistance must be provided.
|
|
3.7
|
Recommended methods and precautions concerning handling, storage, transport or fire
|
|
|
A Safety Data Sheet in accordance with Article 27 of the Directive of 1967 must be provided for all active substances.
|
|
3.8
|
Procedures for destruction or decontamination
|
|
3.8.1
|
Controlled incineration
|
|
|
In many cases the preferred or sole means to safely dispose of active substances, contaminated materials, or contaminated packaging, is through controlled incineration in a licensed incinerator. Where the content of halogens of the active substance is greater than 60%, the pyrolytic behaviour of the active substance under controlled conditions (including, where relevant, supply of oxygen and residence time), at 800°C and the content of polyhalogenated dibenzo-p-dioxins and dibenzo-furans in the products of pyrolysis must be reported. The applicant must provide detailed instructions for safe disposal.
|
|
3.8.2
|
Others
|
|
|
Other methods to dispose of the active substance, contaminated packaging and contaminated materials, where proposed, must be fully described. Data must be provided for such methods, to establish their effectiveness and safety.
|
|
3.9
|
Emergence measures in case of an accident
|
|
|
Procedures for the decontamination of water in case of an accident must be provided.
|
|
| | |
4
|
Analytical methods
|
|
4.1
|
Analytical methods for the determination of pure active substance and, where appropriate, for relevant breakdown products, isomers and impurities of the active substance and additives (e.g. stabilizers)
|
|
4.2
|
Analytical methods including recovery rates and the limits of determination for residues in, and where relevant on, the following:
|
|
4.2.1
|
Treated plants, plant products, foodstuffs, feeding stuffs
|
|
4.2.2
|
Soil
|
|
4.2.3
|
Water (including drinking water)
|
|
4.2.4
|
Air
|
|
4.2.5
|
Animal and human body fluids and tissues
|
|
5
|
Toxicological and metabolism studies on the active substance
|
|
5.1
|
Acute toxicity
|
|
5.1.1
|
Oral
|
|
5.1.2
|
Percutaneous
|
|
5.1.3
|
Inhalation
|
|
5.1.4
|
Intraperitoneal
|
|
5.1.5
|
Skin and where appropriate eye irritation
|
|
5.1.6
|
Skin sensitization
|
|
5.2
|
Short-term toxicity
|
|
5.2.1
|
Oral cumulative toxicity (28-day study)
|
|
5.2.2
|
Oral administration — two species, one rodent (preferably rat) and one non-rodent, usually 90-day study
|
|
5.2.3
|
Other routes (inhalation, percutaneous as appropriate)
|
|
5.3
|
Chronic toxicity
|
|
5.3.1
|
Oral long-term toxicity and carcinogenicity (rat and other mammalian species) — other routes as appropriate
|
|
5.4
|
Mutagenicity — test battery to assess gene mutations, chromosomal aberrations and DNA perturbations
|
|
5.5
|
Reproductive toxicity
|
|
5.5.1
|
Teratogenicity studies — rabbit and one rodent species, oral and when appropriate percutaneous
|
|
5.5.2
|
Multi-generation studies in mammals (at least two generations)
|
|
5.6
|
Metabolism studies in mammals
|
|
5.6.1
|
Absorption, distribution and excretion studies — following both oral and percutaneous administration
|
|
5.6.2
|
Elucidation of metabolic pathways
|
|
5.7
|
Neurotoxicity studies — including where appropriate delayed neurotoxicity tests in adult hens
|
|
5.8
|
Supplementary studies
|
|
5.8.1
|
Toxic effects of metabolites from treated plants in cases where different from those identified in animal studies
|
|
5.8.2
|
Any mechanistic studies needed to clarify effects reported in toxicity studies
|
|
5.9
|
Toxic effects on livestock and pets
|
|
5.10
|
Medical data
|
|
5.10.1
|
Medical surveillance on manufacturing plant personnel
|
|
5.10.2
|
Direct observation, e.g. clinical cases and poisoning incidents
|
|
5.10.3
|
Health records, both from industry and agriculture
|
|
5.10.4
|
Observations on exposure of the general population and epidemiological studies if appropriate
|
|
5.10.5
|
Diagnosis of poisoning (determination of active substance, metabolites), specific signs of poisoning, clinical tests
|
|
5.10.6
|
Sensitization/allergenicity observations
|
|
5.10.7
|
Proposed treatment: first aid measures, antidotes, medical treatment
|
|
5.10.8
|
Prognosis of expected effects of poisoning
|
|
5.11
|
Summary of mammalian toxicology and conclusion (including no observable adverse effect level (NOAEL), no observable effect level (NOEL), acceptable daily intake (ADI). Overall evaluation with regard to all toxicological data, and other information concerning the active substance
|
|
6
|
Residues in or on treated products, food and feed
|
|
6.1
|
Identification of breakdown and reaction products and of metabolites in treated plants or products
|
|
6.2
|
Behaviour of residue of the active substance and its metabolites from the time of application until harvest or out-loading of stored products — uptake and distribution in, and where relevant on, plants, kinetics of disappearance, binding to plant constituents, etc.
|
|
6.3
|
Overall material balance for the active substance. Sufficient residue data from supervised trials to demonstrate that residues likely to arise from the proposed treatments would not be or concern for human and animal health
|
|
6.4
|
Estimation of the potential and actual exposure through diet and other means, such as residue monitoring data for products in the distribution chain, or such as data concerning exposure via air, water, etc.
|
|
6.5
|
Feeding and metabolism studies in livestock (if residues remain in or on crops or parts of crops used for feed) to permit evaluation of residues in foodstuffs of animal origin
|
|
6.6
|
Effects of industrial processing and/or household preparation on the nature and magnitude of residues
|
|
6.7
|
Summary and evaluation of residue behaviour resulting from data submitted pursuant to points 6.1 to 6.6
|
|
7
|
Fate and behaviour in the environment
|
|
7.1
|
Fate and behaviour in soil
|
|
7.1.1
|
Rate and route of degradation (to 90 per cent degradation) including identification of the processes involved and identification of metabolites and breakdown products in at least three soil types under appropriate conditions
|
|
7.1.2
|
Adsorption and desorption in at least three soil types and where relevant adsorption and desorption of metabolites and breakdown products
|
|
7.1.3
|
Mobility in at least three soil types and where relevant mobility of metabolites and breakdown products
|
|
7.1.4
|
Extent and nature of bound residues
|
|
7.2
|
Fate and behaviour in water and air
|
|
7.2.1
|
Rate and route of degradation in aquatic systems — biodegradation, hydrolysis, photolysis (as far as not covered by point 2.8), including identification of metabolites and breakdown products
|
|
7.2.2
|
Adsorption and desorption in water (sedimentation) and where relevant adsorption and desorption of metabolites and breakdown products
|
|
7.2.3
|
Rate and route of degradation in air (for fumigants and other volatile active substances) (as far as not covered by point 2.9)
|
|
8
|
Ecotoxicological studies on the active substance
|
|
8.1
|
Effects on birds
|
|
8.1.1
|
Acute oral toxicity
|
|
8.1.2
|
Short-term toxicity — eight-day dietary study in at least one species (other than chicken)
|
|
8.1.3
|
Effects on reproduction
|
|
8.2
|
Effects on aquatic organisms
|
|
8.2.1
|
Acute toxicity to fish
|
|
8.2.2
|
Chronic toxicity to fish
|
|
8.2.3
|
Effects on fish reproduction and growth rate
|
|
8.2.4
|
Bioaccumulation in fish
|
|
8.2.5
|
Acute toxicity for Daphnia magna
|
|
8.2.6
|
Daphnia magna reproduction and growth rate
|
|
8.2.7
|
Effects on algal growth
|
|
8.3
|
Effects on other non-target organisms
|
|
8.3.1
|
Acute toxicity to honeybees and other beneficial arthropods (e.g. predators)
|
|
8.3.2
|
Toxicity to earthworms and to other soil non-target macro-organisms
|
|
8.3.3
|
Effects on soil non-target micro organisms
|
|
8.3.4
|
Effects on other non-target organisms (flora and fauna) believed to be at risk
|
|
8.3.5
|
Effects on biological methods for sewage treatment
|
|
9
|
Summary and evaluation of points 7 and 8
|
|
10
|
Proposals including justification for the proposals for the classification and labelling of the active substance according to Council Directive 67/548/EEC
|
|
|
— Hazard symbol(s)
|
|
|
— Indications of danger
|
|
|
— Risk phrases
|
|
|
— Safety phrases
|
|
11
|
A dossier as referred to in Annex III, part A, for a representative plant protection product
|
|
| |
PART B
|
| |
Micro-organisms and viruses
|
| |
(this part does not apply to GMOs where points come under Directive 90/220/EEC)
|
| | |
1
|
Identity of the organism
|
|
1.1
|
Applicant (name, address, etc.)
|
|
1.2
|
Manufacturer (name, address, including location of plant)
|
|
1.3
|
Common name or alternative and superseded names
|
|
1.4
|
Taxonomic name and strain for bacteria, protozoa and fungi, indication whether it is a stock variant or a mutant strain; for viruses the taxonomic designation of the agent, serotype, strain or mutant
|
|
1.5
|
Collection and culture reference number where the culture is deposited
|
|
1.6
|
The appropriate test procedures and criteria used for identification (e.g. morphology, biochemistry, serology)
|
|
1.7
|
Composition — microbiological purity, nature, identity, properties, content of any impurities and extraneous organisms
|
|
2
|
Biological properties of the organism
|
|
2.1
|
Target organism. Pathogenicity or kind of antagonism to host, infective dose, transmissibility and information on mode of action
|
|
2.2
|
History of the organism and its uses. Natural occurrence and geographical distribution
|
|
2.3
|
Host specificity range and effects on species other than the target harmful organism including species most closely related to the target species — to include infectivity, pathogenicity and transmissibility
|
|
2.4
|
Infectivity and physical stability when used according to the proposed method. Effect of temperature, exposure to air radiation, etc. Persistence under the likely environmental conditions of use
|
|
2.5
|
Whether the organism is closely related to a plant pathogen or to a pathogen of a vertebrate species or a non-target invertebrate species
|
|
2.6
|
Laboratory evidence of genetic stability (i.e. mutation rate) under environmental conditions of proposed use
|
|
2.7
|
Presence, absence or production of toxins as well as their nature, identity, chemical structure (if appropriate) and stability
|
|
3
|
Further information on the organism
|
|
3.1
|
Function, e.g. fungicide, herbicide, insecticide, repellant, growth regulator
|
|
3.2
|
Effects on harmful organisms, e.g. contact poison, inhalation poison, stomach poison, fungitoxic or fungistatic, etc. systemic or not in plants
|
|
3.3
|
Field of use envisaged, e.g. field, glasshouse, food or feed storage, home garden
|
|
3.4
|
Where necessary, in the light of the test results, any specific agricultural, plant health or environmental conditions under which the active substance may or may not be used
|
|
3.5
|
Harmful organisms controlled and crops or products protected or treated
|
|
3.6
|
Method of production with descriptions of the techniques used to ensure a uniform product and of assay methods for its standardization. In the case of a mutant, detailed information should be provided on its production and isolation, together with all known differences between the mutant and the parent wild strains.
|
|
3.7
|
Methods to prevent loss of virulence of seed stock
|
|
3.8
|
Recommended methods and precautions concerning handling, storage, transport or fire
|
|
3.9
|
Possibility of rendering the organism uninfective
|
|
4
|
Analytical methods
|
|
4.1
|
Methods for establishing the identity and purity of seed stock from which batches are produced and results obtained, including information on variability
|
|
4.2
|
Methods to show microbiological purity of the final product and showing that contaminants have been controlled to an acceptable level, results obtained and information on variability
|
|
4.3
|
Methods used to show that there are no human or other mammalian pathogens as contaminants in the active agent, including in the case of protozoa and fungi, the effects of temperature (35°C and other relevant temperatures)
|
|
4.4
|
Methods to determine viable and non-viable (e.g. toxins) residues in or on treated products, foodstuffs, feeding stuffs, animal and human body fluids and tissues, soil, water and air, where relevant
|
|
5
|
Toxicological, pathogenicity and infectivity studies
|
|
5.1
|
Bacteria, fungi, protozoa and mycoplasma
|
|
5.1.1
|
Toxicity and/or pathogenicity and infectivity
|
|
5.1.1.1
|
Oral single dose
|
|
5.1.1.2
|
In cases where a single dose is not appropriate to assess pathogenicity, a set of range-finding texts must be carried out to reveal highly toxic agents and infectivity
|
|
5.1.1.3
|
Percutaneous single dose
|
|
5.1.1.4
|
Inhalation single dose
|
|
5.1.1.5
|
Intraperitoneal single dose
|
|
5.1.1.6
|
Skin and, where necessary, eye irritation
|
|
5.1.1.7
|
Skin sensitization
|
|
5.1.2
|
Short-term toxicity (90 days exposure)
|
|
5.1.2.1
|
Oral administration
|
|
5.1.2.2
|
Other routes (inhalation, percutaneous as appropriate)
|
|
5.1.3
|
Supplementary toxicological and/or pathogenicity and infectivity studies
|
|
5.1.3.1
|
Oral long-term toxicity and carcinogenicity
|
|
5.1.3.2
|
Mutagenicity — (tests as referred to under point 5.4 of part A)
|
|
5.1.3.3
|
Teratogenicity studies
|
|
5.1.3.4
|
Multi-generation study in mammals (at least two generations)
|
|
5.1.3.5
|
Metabolic studies — absorption, distribution and excretion in mammals including elucidation of metabolic pathways
|
|
5.1.3.6
|
Neurotoxicity studies, including where appropriate delayed neurotoxicity tests in adult hens
|
|
5.1.3.7
|
Immunotoxicity, e.g. allergenicity
|
|
5.1.3.8
|
Pathogenicity and infectivity under immunosuppression
|
|
5.2
|
Viruses, viroids
|
|
5.2.1
|
Acute toxicity and/or pathogenicity and infectivity
|
|
|
Data as outlined under point 5.1.1 and cell culture studies using purified infective virus and primary cell cultures of mammalian, avian and fish cells
|
|
5.2.2
|
Short-term toxicity
|
|
|
Data as outlined under point 5.1.2 and tests for infectivity carried out by bio-assay or on a suitable cell culture at least seven days after the last administration to the test animals
|
|
5.2.3
|
Supplementary toxicological and/or pathogenicity and infectivity studies as outlined under point 5.1.3.
|
|
5.3
|
Toxic effects on livestock and pets
|
|
5.4
|
Medical data
|
|
5.4.1
|
Medical surveillance on manufacturing plant personnel
|
|
5.4.2
|
Health records, both from industry and agriculture
|
|
5.4.3
|
Observations on exposure of the general population and epidemiological data, if appropriate
|
|
5.4.4
|
Diagnosis of poisoning, specific signs of poisoning, clinical tests, if appropriate
|
|
5.4.5
|
Sensitization/allergenicity observations, if appropriate
|
|
5.4.6
|
Proposed treatment: first aid measures, antidotes, medical treatment, if appropriate
|
|
5.4.7
|
Prognosis of expected effects of poisoning, if appropriate
|
|
5.5
|
Summary of mammalian toxicology and conclusions (including NOAEL, NOEL and ADI, if appropriate). Overall evaluation with regard to all toxicological pathogenicity and infectivity data, and infectivity and other information concerning the active substance
|
|
6
|
Residues in or on treated products, food and feed
|
|
6.1
|
Identification of viable and non-viable (e.g. toxins) residues in or on treated plants or products, the viable residue by culture or bio-assay and the non-viable by appropriate techniques
|
|
6.2
|
Likelihood of multiplication of the active substance in or on crops or food together with a report on any effect on food quality
|
|
6.3
|
In cases where residues of toxins remain in or on an edible plant product, data as outlined under points 4.2.1 and 6 of part A are required
|
|
6.4
|
Summary and evaluation of residue behaviour resulting from data submitted under points 6.1 to 6.3
|
|
7
|
Fate and behaviour to the environment
|
|
7.1
|
Spread, mobility, multiplication and persistence in air, water, soil
|
|
7.2
|
Information concerning possible fate in food chains
|
|
7.3
|
In cases where toxins are produced, data as outlined under part A, point 7 are required, where relevant
|
|
8
|
Ecotoxicological studies
|
|
8.1
|
Birds — acute oral toxicity and/or pathogenicity and infectivity
|
|
8.2
|
Fish — acute toxicity and/or pathogenicity and infectivity
|
|
8.3
|
Toxicity — Daphnia magna (if appropriate)
|
|
8.4
|
Effects on algal growth
|
|
8.5
|
Important parasites and predators of target species; acute toxicity and/or pathogenicity and infectivity
|
|
8.6
|
Honey bees: acute toxicity and/or pathogenicity and infectivity
|
|
8.7
|
Earthworms: acute toxicity and/or pathogenicity and infectivity
|
|
8.8
|
Other non-target organisms believed to be at risk: acute toxicity and/or pathogenicity and infectivity
|
|
8.9
|
Extent of indirect contamination on adjacent non-target crops, wild plants, soil and water
|
|
8.10
|
Effects on other flora and fauna
|
|
8.11
|
In cases where toxins are produced, data as outlined under Part A, points 8.1.2, 8.1.3, 8.2.2, 8.2.3, 8.2.4, 8.2.5, 8.2.6, 8.2.7 and 8.3.3 are required, where relevant
|
|
9
|
Summary and evaluation of points 7 and 8
|
|
10
|
Proposals including justification of the proposals for the classification and labelling of the active substance in accordance with Directive 67/548/EEC
|
|
|
— Hazard symbol(s)
|
|
|
— Indications of danger
|
|
|
— Risk phrases
|
|
|
— Safety phrases
|
|
11
|
A dossier referred to in Annex III, part B, for a representative plant protection product
|
|
| |
Part 2
|
| |
Annex III
|
| |
(Annex III to the Directive of 1991, as amended by Commission Directive No 93/71/EEC of 27 July 1993)
|
| |
REQUIREMENTS FOR THE DOSSIER TO BE SUBMITTED FOR THE AUTHORIZATION OF A PLANT PROTECTION PRODUCT
|
| |
INTRODUCTION
|
| |
The information required shall:
|
| | |
1.1
|
include a technical dossier supplying the information necessary for evaluating efficacy and the foreseeable risks, whether immediate or delayed, which the plant protection product may entail for humans, animals and the environment and containing at least the information and results of the studies referred to below;
|
|
1.2
|
where relevant, be generated using test guidelines referred to or described in this Annex; in the case of studies initiated before the adoption of the modification of this Annex, the information shall be generated using suitable internationally or nationally validated test guidelines or, in the absence thereof, test guidelines accepted by the competent authority;
|
|
1.3
|
in the event of a test guideline being inappropriate or not described, or where another one than those referred to in this annex has been used, include a justification, which is acceptable to the competent authority for the gidelines used;
|
|
1.4
|
include, a full description of test guidelines used, except if they are referred to or described in this Annex, and a full description of any deviations from them including a justification, which is acceptable to the competent authority, for these deviations;
|
|
1.5
|
include a full and unbiased report of the studies conducted as well as a full description of them or a justification, which is acceptable to the competent authority where:
|
|
|
— particular data and information which would not be necessary owing to the nature of the product or its proposed uses, are not provided, or
|
|
|
— it is not scientifically necessary, or technically possible to supply information and data.
|
|
1.6
|
where relevant, have been generated in accordance with the requirements of Directive 86/609/EEC.
|
|
2.1
|
Tests and analyses must be conducted in accordance with the principles laid down in Directive 87/18/EEC, where testing is done to obtain data on the properties and/or safety with respect to human health or the environment.
|
|
2.2
|
Tests and analyses, required under the provisions of section 6 points 6.2 to 6.6 of this annex, shall, where they are conducted outside the territory of the state, be conducted by official or officially recognized testing facilities or organisations in the Member State concerned, which satisfy at least the requirements specified in points 2.2 and 2.3 of the introduction to Annex III to Directive 93/71/EEC.
|
|
2.3
|
Tests and analyses, required under the provisions of section 6 points 6.2 to 6.6 of this annex, shall, where they are conducted within the territory of the state, be conducted in accordance with the Principles of Good Experimental Practice set out in the Sixth Schedule, or in compliance with Irish/European Standard IS/EN 45001 and in accordance with the authorization for trials or trials permit concerned.
|
|
2.4
|
Notwithstanding the provisions of point 2.1, during the period to 31 December 1999, tests and analyses done to obtain data on the properties and/or safety with respect to honeybees and beneficial arthropods other than bees may have been conducted by officially recognized testing facilities or organisations, in accordance with the principles laid down in the Sixth Schedule, or in compliance with Irish/European Standard IS/EN 45001, where they are conducted within the territory of the state, and in accordance with the requirements of points 2.2 and 2.3 of the introduction to Annex III to Directive 93/71/EEC, where they are conducted outside the territory of the state.
|
|
3
|
The information required shall include the proposed classification and labelling of the plant protection product in accordance with relevant Community Directives.
|
|
4
|
In individual cases it may be necessary to require certain information as provided for in Annex II, Part A, for formulants. Before such information will be required and before possibly new studies have to be performed, all information on the formulant, made available to the competent authority, shall be considered, in particular when:
|
|
|
— the use of the formulant is permitted in food, animal feeding stuffs, medicines or cosmetics in accordance with Community legislation; or
|
|
|
— a safety data sheet has been submitted for the formulant in accordance with Council Directive 67/548/EEC of 27 June 1967, on the approximation of laws, regulations and administrative provisions relating to the classification, packaging and labelling of dangerous substances.
|
|
| |
PART A
|
| |
Chemical preparations
|
| | |
1
|
Identity of the plant protection product
|
|
|
The information provided, taken together with that provided for the active substance(s), must be sufficient to identify preparations, with precision, and to define them in terms of their specification and nature. The information and data referred to, unless otherwise specified, are required for all plant protection products.
|
|
1.1
|
Applicant (name and address, etc.)
|
|
|
The name and address of the applicant (permanent community address) must be provided as must the name, position, telephone and telefax number of the appropriate person to contact.
|
|
|
Where, in addition, the applicant has an office, agent or representative in the territory of the State, the name and address of the local office, agent or representative should be provided, as should the name, position, telephone and telefax number of the appropriate person to contact.
|
|
1.2
|
Manufacturer of the preparation and the active substance(s) (names and addresses, etc. including location of plants)
|
|
|
The name and address of the manufacturer of the preparation and of each active substance in the preparation must be provided as must the name and address of each manufacturing plant in which the preparation and active substance are manufactured. A contact point (preferably a central contact point, to include name, telephone and telefax numbers) must be provided for each.
|
|
|
If the active substance originates from a manufacturer from which data according to Annex II had not been submitted previously, a statement of purity and detailed information on the impurities as required in Annex II must be provided.
|
|
1.3
|
Trade name or proposed trade name, and manufacturer's development code number of the preparation if appropriate
|
|
|
All former and current trade names and proposed trade names and development code numbers of the preparation as well as the current names and numbers must be provided. Where trade names and code numbers referred to, relate to similar but different preparations (possibly obsolete), full details of the differences, must be provided. (The proposed trade name may not give rise to confusion with the trade name of plant protection products already authorized).
|
|
1.4
|
Detailed quantitative and qualitative information on the composition of the preparation (active substance(s), and formulant (s)
|
|
1.4.1
|
For preparations the following information must be reported—
|
|
|
— the content of both technical active substance(s) and pure active substance(s); and
|
|
|
— the content of formulants,
|
|
|
The concentrations must be expressed in terms as provided for in Article 6(2) of the Directive of 197.
|
|
1.4.2
|
For active substances their ISO common names or proposed ISO common names and their CIPAC numbers, and, where available, the EEC (EINECS or ELINCS) numbers must be provided. Where relevant it must be stated which salt, ester, anion or cation is present.
|
|
1.4.3
|
Formulants must where possible, be identified both by their chemical name as given in Annex I to the Directive of 1967, or, if not included in that Directive, in accordance with both IUPAC and CA nomenclature. Their structure or structural formula must be provided. For each component of formulants the relevant EEC (EINECS or ELINCS) number and CAS number where they exist, must be provided. Where the information provided does not fully identify a formulant, an appropriate specification must be provided. The trade name of formulants, where they exist, must also be provided.
|
|
1.4.4
|
For formulants the function must be given:
|
|
|
adhesive (sticker)
|
preservative
|
|
|
antifoaming agent
|
odourant
|
|
|
antifreeze
|
perfume
|
|
|
binder
|
propellant
|
|
|
buffer
|
repellent
|
|
|
carrier
|
safener
|
|
|
deodorant
|
solvent
|
|
|
dispersing agent
|
stabiliser
|
|
|
dye
|
synergist
|
|
|
emetic
|
thickener
|
|
|
emulsifier
|
wetting agent
|
|
|
fertilizer
|
miscellaneous (specify)
|
|
1.5
|
Physical state and nature of the preparation (emulsifiable concentrate, wettable powder, solution etc.)
|
|
1.5.1
|
The type and code of preparation must be designated in accordance with Annex 2 to Part 2 of the Fourth Schedule.
|
|
|
Where a particular preparation is not defined precisely in that Annex, a full description of the physical nature and state of the preparation must be provided, together with a proposal for a suitable description of the type of preparation and a proposal for its definition.
|
|
1.6
|
Function (herbicide, insecticide, etc.)
|
|
|
The function must be specified from among the following:
|
|
|
acaricide
|
plant growth regulator
|
|
|
bactericide
|
repellant
|
|
|
fungicide
|
rodenticide
|
|
|
herbicide
|
semio-chemicals
|
|
|
insecticide
|
talpicide
|
|
|
molluscicide
|
viricide
|
|
|
nematicide
|
other (must be specified)
|
|
2
|
Physical, chemical and technical properties of the plant protection product
|
|
|
The extent to which plant protection products for which authorization is sought, comply with relevant FAO specifications as agreed by the Group of Experts on Pesticide Specifications, of the FAO Panel of Experts on Pesticide Specifications, Registration Requirements and Application Standards, must be stated. Divergences from FAO specifications must be described in detail, and justified.
|
|
2.1
|
Appearance (colour and odour)
|
|
|
A description of both the colour and odour, if any, and the physical state of the preparation, must be provided.
|
|
2.2
|
Explosivity and oxidizing properties
|
|
2.2.1
|
The explosive properties of preparations must be determined in accordance with EEC Method A 14 and be reported. Where available thermodynamic information establishes beyond reasonable doubt, that the preparation is incapable of exothermic reaction, it is sufficient to provide that information as a justification for not determining the explosive properties of the preparation.
|
|
2.2.2
|
The oxidizing properties of preparations which are solids, must be determined in accordance with EEC Method A 17 and be reported. For other preparations the method used must be justified. Oxidizing properties do not have to be determined if it can be shown without reasonable doubt, on the basis of thermodynamic information, that the preparation is incapable of reacting exothermically with combustible materials.
|
|
2.3
|
Flash point and other indications of flammability or spontaneous ignition
|
|
|
The flash point of liquids which contain flammable solvents, must be determined in accordance with EEC Method A 9 and be reported. The flammability of solid preparations and gasses must be determined in accordance with EEC Method A 10, A 11 or A 12, as appropriate, and be reported. The auto-flammability of preparations, determined in accordance with EEC Method A 15 or A 16 as appropriate, and/or, where necessary, in accordance with the UN-Bowes-Cameron-Cage-Test (UN-Recommendations on the Transport of Dangerous Goods, Chapter 14, Nr. l4.3.4), must be reported.
|
|
2.4
|
Acidity/alkalinity and if necessary pH value
|
|
2.4.1
|
In the case of preparations which are acidic (pH10) the acidity or alkalinity and the pH value must be determined in accordance with CIPAC Method MT31 and MT75 respectively, and be reported.
|
|
2.4.2
|
Where relevant (if to be applied as aqueous dilution) the pH of a 1% aqueous dilution, emulsion or dispersion of the preparation, must be determined in accordance with CIPAC Method MT75 and be reported.
|
|
2.5
|
Viscosity and surface tension
|
|
2.5.1
|
In the case of liquid preparations for Ultra Low Volume use (ULV) the kinematic viscosity must be determined in accordance with OECD Test Guideline 14 and be reported.
|
|
2.5.2
|
For non newtonian liquids the viscosity must be determined and reported together with the test conditions.
|
|
2.5.3
|
In the case of liquid preparations the surface tension must be determined in accordance with EEC Method A 5 and be reported,
|
|
2.6
|
Relative density and bulk density
|
|
2.6.1
|
The relative density of liquid preparations must be determined in accordance with EEC Method A 3 and be reported.
|
|
2.6.2
|
The bulk (tap) density of preparations which are powders or granules, must be determined in accordance with CIPAC Methods MT33, MT159 or MT169, as appropriate, and be reported.
|
|
2.7
|
Storage stability — stability and shelf-life. Effects of light, temperature and humidity on technical characteristics of the plant protection product
|
|
2.7.1
|
The stability of the preparation after storage for 14 days at 54°C must be determined in accordance with CIPAC Method MT46 and be reported. Other storage times and/or temperatures may be needed (e.g. 8 weeks at 40°C or 12 weeks at 35°C or 18 weeks at 30°C) if the preparation is heat sensitive.
|
|
|
If the active substance content after the heat stability test has decreased by more than 5% of the initial determined content, the minimum content must be declared and information on the degradation products must be supplied.
|
|
2.7.2
|
Additionally in the case of liquid preparations, the effect of low temperatures on stability, must be determined in accordance with CIPAC Methods MT39, MT48, MT51 or MT54, as appropriate, and be reported.
|
|
2.7.3
|
The shelf life of the preparation at ambient temperatures must be reported. Where shelf life is less than two years, the shelf life in months, with appropriate temperature specifications, must be reported. Useful information relating to such testing is contained in GIFAP Monograph No 17.
|
|
2.8
|
Technical characteristics of the plant protection product
|
|
|
The technical characteristics of the preparation must be determined to permit a decision to be made as to its acceptability.
|
|
2.8.1
|
Wettability
|
|
|
The wettability of solid preparations which are diluted for use (e.g. wettable powders, water soluble powders, water soluble granules and water dispersible granules), must be determined in accordance with CIPAC Method MT53.3 and be reported.
|
|
2.8.2
|
Persistent foaming
|
|
|
The persistence of foaming of preparations to be diluted with water, must be determined in accordance with CIPAC Method MT47 must be reported.
|
|
2.8.3
|
Suspensibility and suspension stability
|
|
2.8.3.1
|
The suspensibility of water dispersible products (e.g. wettable powders, water dispersible granules, suspension concentrates) must be determined in accordance with CIPAC Method MT15, MT161 or MT168, as appropriate and be reported.
|
|
2.8.3.2
|
The spontaneity or dispersibility of water dispersible products (e.g. suspension concentrates and water dispersible granules) must be determined in accordance with CIPAC Methods MT160 or MT174, as appropriate, and be reported.
|
|
2.8.4
|
Dilution stability
|
|
|
The dilution stability of water soluble products must be determined in accordance with CIPAC Method MT41 and be reported.
|
|
2.8.5
|
Dry sieve test and wet sieve test
|
|
|
In order to ensure that dustable powders have a suitable particle size distribution for ease of application, a dry sieve test must be conducted in accordance with CIPAC Method MT59.1 and be reported.
|
|
|
In the case of water dispersible products, a wet sieve test must be conducted in accordance with CIPAC Method MT59.3 or MT167, as appropriate, must be reported.
|
|
2.8.6
|
Particle size distribution (dustable and wettable powders, granules), content of dust/fines (granules), attrition and friability (granules)
|
|
2.8.6.1
|
The size distribution of particles in the case of powders, must be determined in accordance with OECD Method 110 and be reported.
|
|
|
The nominal size range of granules for direct application must be determined in accordance with CIPAC MT58.3 and for water dispersible granules and in accordance with CIPAC MT170, and be reported.
|
|
2.8.6.2
|
The dust content of granular preparations, must be determined in accordance with CIPAC Method MT171 and be reported. If relevant for operator exposure the particle size of dust must be determined in accordance with OECD Method 110 and be reported.
|
|
2.8.6.3
|
The friability and attrition characteristics of granules, must be determined and reported once internationally agreed methods are available. Where relevant data are available they must be reported together with details of the method used.
|
|
2.8.7
|
Emulsifiability, Re-emulsifiability, emulsion stability
|
|
2.8.7.1
|
The emulsifiability, emulsion stability and re-emulsifiability of preparations which form emulsions, must be determined in accordance with CIPAC Methods MT36 or MT173, as appropriate, and be reported.
|
|
2.8.7.2
|
The stability of dilute emulsions and of preparations which are emulsions, must be determined in accordance with CIPAC Method MT20 or MT173, as appropriate, and be reported.
|
|
2.8.8
|
Flowability, pourability (rinsability) and dustability
|
|
2.8.8.1
|
The flowability of granular preparations must be determined in accordance with CIPAC Method MT172 and be reported.
|
|
2.8.8.2
|
The pourability (including rinsed residue) of suspensions (e.g. suspension concentrates, suspo-emulsions), must be determined in accordance with CIPAC Method MT148 and be reported.
|
|
2.8.8.3
|
The dustability of dustable powders following accelerated storage as specified in paragraph 2.7.1 must be determined in accordance with CIPAC Method MT34 or another suitable method and be reported.
|
|
2.9
|
Physical and chemical compatibility with other products including plant protection products with which its use is to be authorized
|
|
2.9.1
|
The physical compatibility of tank mixes must be determined using in-house test methods and be reported. A practical test is an acceptable alternative.
|
|
2.9.2
|
The chemical compatibility of tank mixes must be determined and reported except where examination of the individual properties of the preparations establishes beyond reasonable doubt that there is no possibility of reaction taking place. In such cases it is sufficient to provide that information as justification for not determining chemical compatibility.
|
|
2.10
|
Adherence and distribution to seeds
|
|
|
In the case of preparations for seed treatment, both distribution and adhesion must be investigated and reported; in the case of distribution in accordance with CIPAC Method MT175.
|
|
2.1.1
|
Summary and evaluation of data presented under points 2.1 to 2.10
|
|
3
|
Data on application
|
|
3.1
|
Field of use envisaged, e.g. field, protected crops, storage of plant products, home gardening
|
|
|
The field(s) of use, existing and proposed, for preparations containing the active substance must be specified from among the following:
|
|
|
Field use
|
— Agriculture
|
|
|
|
— Horticulture
|
|
|
|
— Forestry
|
|
|
|
— Viticulture
|
|
|
Protected crops
|
|
|
Amenity
|
|
|
Weed control on non-cultivated areas
|
|
|
Home gardening
|
|
|
House plants
|
|
|
Plant products storage practice
|
|
|
Other (specify)
|
|
3.2
|
Effects on harmful organisms, e.g. contact, inhalation or stomach poison, fungitoxic or fungistatic, etc., systemic or not in plants
|
|
3.2.1
|
The nature of the effects on harmful organisms must be stated:
|
|
|
contact action
|
|
|
stomach action
|
|
|
inhalation action
|
|
|
fungitoxic action
|
|
|
fungistatic action
|
|
|
desiccant
|
|
|
reproduction inhibitor
|
|
|
other (must be specified)
|
|
|
It must be stated whether or not the active substance is translocated in plants and where relevant whether such translocation is apoplastic, symplastic or both.
|
|
3.3
|
Details of intended use e.g. types of harmful organisms controlled and/or plants or plant products to be protected
|
|
|
Details of the intended use must be provided.
|
|
|
Where relevant, effects achieved e.g. sprout suppression, retardation of ripening, reduction in stem length, enhanced fertilization etc. must be reported.
|
|
3.4
|
Application rate
|
|
|
For each method of application and each use, the rate of application per unit (ha, m , m3) treated, in terms of g or kg of both preparation and active substance, must be provided.
|
|
|
Application rates shall normally be expressed in g or kg/ha or in kg/m3 and where appropriate in g or kg/tonne; for protected crops and home gardening use rates shall be expressed in g or kg/100mor g or kg/m3.
|
|
3.5
|
Concentration of active substance in material used (e.g. in the diluted spray, baits or treated seed)
|
|
|
The content of active substance shall be reported, as appropriate, in g/l, g/kg, mg/kg or in g/tonne.
|
|
3.6
|
Method of application
|
|
|
The method of application proposed must be described fully, indicating the type of equipment to be used, if any, as well as the type and volume of diluent to be used per unit of area or volume.
|
|
3.7
|
Number and timing of applications and duration of protection
|
|
|
The maximum number of applications to be used and their timing, must be reported. Where relevant the growth stages of the crop or plants to be protected and the development stages of the harmful organisms, must be indicated. Where possible the interval between applications, in days, must be stated.
|
|
|
The duration of protection afforded both by each application and by the maximum number of applications to be used, must be indicated.
|
|
3.8
|
Necessary waiting periods or other precautions to avoid Phytotoxic effects on succeeding crops
|
|
|
Where relevant, minimum waiting periods between last application and sowing or planting of succeeding crops, which are necessary to avoid phytotoxic effects on succeeding crops, must be stated, and follow from the data provided under paragraph 6.6.
|
|
|
Limitations on choice of succeeding crops, if any, must be stated.
|
|
3.9
|
Proposed instructions for use
|
|
|
The proposed instructions for use of the preparation, to be printed on labels and leaflets, must be provided.
|
|
4
|
Further information on the plant protection product
|
|
4.1
|
Packaging (type, materials, size etc.), compatibility of the preparation with proposed packaging materials
|
|
4.1.1
|
Packaging to be used must be fully described and specified in terms of the materials used, manner of construction (e.g. extruded, welded etc.), size and capacity, size of opening, type of closure and seals. It must be designed in accordance with the criteria and guidelines specified in the FAO "Guidelines for the Packaging of Pesticides".
|
|
4.1.2
|
The suitability of the packaging, including closures, in terms of its strength, leakproofness and resistance to normal transport and handling, must be determined in accordance with ADR Methods 3552, 3553, 3560, 3554, 3555, 3556 and 3558, or ADR methods for intermediate bulk containers, as appropriate, and where child resistant closures are required, in accordance with ISO Standard 8317, and be reported.
|
|
4.1.3
|
The resistance of the packaging material to its contents must be determined in accordance with GIFAP Monograph no. 17, and be reported.
|
|
4.2
|
Procedures for cleaning application equipment
|
|
|
Cleaning procedures for both application equipment and protective clothing must be described in detail. The effectiveness of the cleaning procedure, must be fully investigated and reported.
|
|
4.3
|
Re-entry periods, necessary waiting periods or other precautions to protect man, livestock and the environment
|
|
|
The information provided must follow from and be supported by the data provided for the active substance(s) and that provided under sections 7 and 8.
|
|
4.3.1
|
Where relevant, pre-harvest intervals, re-entry periods or withholding periods necessary to minimize the presence of residues in or on crops, plants and plant products, or in or on treated areas or spaces, with a view to protecting man or livestock, must be specified e.g.
|
|
|
— pre-harvest interval (in days) for each relevant crop;
|
|
|
— re-entry period (in days) for livestock, to areas to be grazed;
|
|
|
— re-entry period (in hours or days) for man to crops, buildings or spaces treated;
|
|
|
— withholding period (in days) for animal feedingstuffs;
|
|
|
— waiting period (in days), between application and handling treated products; or
|
|
|
— waiting period (in days), between last application and sowing or planting succeeding crops.
|
|
4.3.2
|
Where necessary, in the light of test results, information on any specific agricultural, plant health or environmental conditions under which the preparation may or may not be used, must be provided.
|
|
4.4
|
Recommended methods and precautions concerning: handling, storage, transport or fire
|
|
|
The recommended methods and precautions concerning handling procedures (detailed) for the storage, at both warehouse and user level of plant protection products, for their transport and in the event of fire must be provided, Where available, information on combustion products must be provided. The risks likely to arise and the methods and procedures to minimize the hazards arising, must be specified. Procedures to preclude or minimize the generation of waste or leftovers must be provided.
|
|
|
Where relevant, assessment must be conducted in accordance with ISO-TR 9122.
|
|
|
Where relevant, the nature and characteristics of protective clothing and equipment proposed must be reported. The data provided must be sufficient to permit an evaluation to be made of the suitability and effectiveness of the protective clothing under realistic conditions of use (e.g. field or glasshouse circumstances).
|
|
4.5
|
Emergency measures in the case of an accident
|
|
|
Whether arising during transport, storage or use, detailed procedures to be followed in the event of an emergency, must be provided, and include procedures for—
|
|
|
— containment of spillages;
|
|
|
— decontamination of areas, vehicles and buildings;
|
|
|
— disposal of damaged packaging, adsorbents and other materials;
|
|
|
— protection of emergency workers and bystanders; and
|
|
|
— first aid measures.
|
|
4.6
|
Procedures for destruction or decontamination of the plant protection product and its packaging
|
|
|
Procedures for destruction and decontamination must be developed for both small quantities (user level) and large quantities (warehouse level). The procedures must be consistent with provisions in place relating to the disposal of waste and of toxic waste. The means of disposal proposed should be without unacceptable influence on the environment and be the most cost effective and practical means of disposal feasible.
|
|
4.6.1
|
Possibility of neutralization
|
|
|
Neutralization procedures (e.g. by reaction with alkali to form less toxic compounds) for use in the event of accidental spillages, must where they are feasible, be described. The products produced after neutralization should be identified (through analysis or on the basis of theoretical considerations)and be reported.
|
|
4.6.2
|
Controlled incineration
|
|
|
In many cases the preferred or sole means to safely dispose of active substances as well as plant protection products containing them, contaminated materials, or contaminated packaging, is through controlled incineration in a licensed incinerator.
|
|
|
Where the content of halogens of the active substance(s) in the preparation is greater than 60%, the pyrolytic behaviour of the active substance under controlled conditions (including, where relevant, supply of oxygen and residence time), at 800°C and the content of polyhalogenated dibenzo-p-dioxins and dibenzo-furans in the products of pyrolysis must be reported. The applicant must provide detailed instructions for safe disposal.
|
|
4.6.3
|
Others
|
|
|
Other methods to dispose of plant protection products, packaging and contaminated materials, where proposed, must be fully described. Data must be provided for such methods, to establish their effectiveness and safety.
|
|
5
|
Analytical methods
|
|
5.1
|
Analytical methods for determining the composition of the plant protection product
|
|
5.2
|
In so far as not covered by Annex II, Part A, point 4.2, analytical methods including recovery rates and the limits of determination for residues and where relevant on, the following;
|
|
5.2.1
|
Treated plants, plant products, foodstuffs, feeding-stuffs
|
|
5.2.2
|
Soil
|
|
5.2.3
|
Water (including drinking water)
|
|
5.2.4
|
Air
|
|
5.2.5
|
Animal and human body fluids and tissues
|
|
6
|
Efficacy data
|
|
|
General
|
|
|
The data supplied must be sufficient to permit an evaluation of the plant protection product to be made. In particular it must be possible to evaluate the nature and extent of benefits that accrue following use of the preparation, where they exist in comparison to suitable reference products and damage thresholds, and to define its conditions of use.
|
|
|
The number of trials to be conducted and reported depends mainly on factors such as the extent to which the properties of the active substance(s) it contains are known and on the range of conditions that arise, including variability in plant health conditions, climatic differences, the range of agricultural practices, the uniformity of the crops, the mode of application, the type of harmful organism and the type of plant protection product.
|
|
|
Sufficient data must be generated and submitted to confirm that patterns determined hold for the regions and the range of conditions, likely to be encountered in the regions concerned, for which its use is to be recommended. Where an applicant claims that tests in one or more of the proposed regions of use are unnecessary because conditions are comparable with those in other regions where tests have been carried out, the applicant must substantiate the claim for comparability with documentary evidence.
|
|
|
In order to assess seasonal differences, if any, sufficient data must be generated and submitted to confirm the performance of the plant protection products in each agronomically and climatically different region for each particular crop (or commodity)/ harmful organism combination. Normally trials on effectiveness or phytotoxicity, where relevant, in at least two growing seasons must be reported.
|
|
|
If in the opinion of the applicant the trials from the first season adequately confirm the validity of claims made on the basis of extrapolation of results from other crops, commodities or situations or from tests with closely similar preparations, a justification, which is acceptable to the competent authority for not carrying out a second seasons's work must be provided. Conversely, where, because of climatic or plant health conditions or other reasons, the data obtained in any particular season are of limited value for the assessment of performance, trials in one or more further seasons must be conducted and reported.
|
|
6.1
|
Preliminary tests
|
|
|
Reports in summary form of preliminary tests, including glasshouse and field studies, used to assess the biological activity and dose range finding of the plant protection product and of the active substance(s) it contains, must be submitted when requested by the competent authority. These reports will provide additional information for the competent authority when it evaluates the plant protection product. Where this information is not submitted a justification which is acceptable to the competent authority must be provided.
|
|
6.2
|
Testing effectiveness
|
|
|
Aim of the tests
|
|
|
The tests shall provide sufficient data to permit an evaluation of the level, duration and consistency of control or protection or other intended effects of the plant protection product in comparison to suitable reference products, where they exist.
|
|
|
Test conditions
|
|
|
Normally a trial consists of three components: test product, reference product and untreated control.
|
|
|
The performance of the plant protection product must be investigated in relation to suitable reference products, where they exist. A suitable reference product is defined as an authorized plant protection product which has proved to have a sufficient performance in practice under the agricultural, plant health and environmental (including climatic) conditions in the area of proposed use. In general, formulation type, effects on the harmful organisms, working spectrum and method of application should be close to those of the tested plant protection product.
|
|
|
Plant protection products must be tested in circumstances where the target harmful organism has been shown to have been present at a level causing or known to cause adverse effects (yield, quality, operational benefit) on an unprotected crop or area or on plants or plant products which have not been treated or where the harmful organism is present at such a level that an evaluation of the plant protection product can be made.
|
|
|
Trials to provide data on plant protection products for control of harmful organisms must show the level of control of the species of harmful organisms concerned or of species representative of groups for which claims are made. Trials must include the different stages of growth or life cycle of the harmful species, where this is relevant and the different strains or races, where these are likely to show different degrees of susceptibility.
|
|
|
Similarly, trials to provide data on plant protection products which are plant growth regulators, must show the level of effects on the species to be treated, and include investigation of differences in the response of a representative sample of the range of cultivars on which its use is proposed.
|
|
|
In order to clarify the dose response, dose rates lower than the recommended one must be included in some trials in order to enable to assess whether the recommended rate is the minimum necessary to achieve the desired effect.
|
|
|
The duration of the effects of treatment must be investigated in relation to the control of the target organism or effect on the treated plants or plant products, as appropriate. When more than one application is recommended, trials must be reported which establish the duration of the effects of an application, the number of applications necessary and the desired intervals between them.
|
|
|
Evidence must be submitted to show that the dose, timing and method of application recommended give adequate control, protection or have the intended effect in the range of circumstances likely to be encountered in practical use.
|
|
|
Unless there are clear indications that the performance of the plant protection product is unlikely to be affected to a significant degree by environmental factors, such as temperature or rain, an investigation of the effects of such factors on performance must be carried out and reported, particularly where it is known that the performance of chemically related products is so affected.
|
|
|
Where proposed label claims include recommendations for the use of the plant protection product with other plant protection product(s) or adjuvant(s) information on the performance of the mixture must be provided.
|
|
|
Test guideline
|
|
|
Trials must be designed to investigate specified issues, to minimize the effects of random variation between different parts of each site and to enable statistical analysis to be applied to results amenable to such analysis. The design, analysis and reporting of trials must be in accordance with European and Mediterranean Plant Protection Organisation (EPPO) guidelines 152 and 181. The report shall include a detailed and critical assessment of the data.
|
|
|
When conducted within the territory of the state, the trials must be carried out in accordance with specific EPPO guidelines, where available. When conducted within the territory of another Member State, the trials must be carried out in accordance with specific EPPO guidelines, where available or when the Member State concerned so requires, in accordance with guidelines satisfying at least the requirements of the corresponding EPPO guidelines.
|
|
|
A statistical analysis of results amenable to such analysis must be carried out; where necessary the test guideline used must be adapted to enable such analysis.
|
|
6.3
|
Information on the occurrence or possible occurrence of the development of resistance
|
|
|
Laboratory data and where it exists, field information relating to the occurrence and development of resistance or cross-resistance in populations of harmful organisms to the active substance(s), or to related active substances, must be provided. Where such information is not directly relevant to the uses for which authorization is sought or to be renewed (different species of harmful organism or different crops), it must, if available, nevertheless be provided, as it may provide an indication of the likelihood of resistance developing in the target population.
|
|
|
Where there is evidence or information to suggest that, in commercial use, the development of resistance is likely, evidence must be generated and submitted as to the sensitivity of the population of the harmful organism concerned to the plant protection product. In such cases a management strategy designed to minimize the likelihood of resistance of cross-resistance developing in target species must be provided.
|
|
6.4
|
Effects on the yield of treated plants or plant products in terms of quantity and/or quality
|
|
6.4.1
|
Effects on the quality of plants or plant products
|
|
|
Aim of the tests
|
|
|
The tests shall provide sufficient data to permit an evaluation of the possible occurrence of taint or odour or other quality aspects of plants or plant products after treatment with the plant protection product.
|
|
|
Circumstances in which required
|
|
|
The possibility of the occurrence of taint or odour in food crops must be investigated and be reported where:
|
|
|
— the nature of the product or its use is such that a risk of occurrence of taint or odour might be expected, or
|
|
|
— other products based on the same or a closely similar active substance have been shown to present a risk of occurrence of taint or odour.
|
|
|
The effects of plant protection products on other quality aspects of treated plants or plant products must be investigated and reported where:
|
|
|
— the nature of the plant protection product or its use could have an adverse influence on other quality aspects (for example in the case of use of plant growth regulators close to harvest), or
|
|
|
— other products based on the same or a closely similar active substance have been shown to have an adverse influence on the quality.
|
|
|
Testing should be conducted initially on the main crops on which the plant protection product is to be used, at twice the normal rates of application and using, where relevant, the main methods of processing, where effects are observed it is necessary to perform testing at the normal rate of application.
|
|
|
The extent of investigation necessary on other crops will depend on their degree of similarity to the main crops already tested, the quantity and quality of data available on those main crops and how far the manner of use of the plant protection product and methods of processing the crops, if relevant, are similar. it is generally sufficient to perform the test with the main formulation type to be authorized.
|
|
6.4.2
|
Effects on transformation processes
|
|
|
Aim of the tests
|
|
|
The tests shall provide sufficient data to permit an evaluation of the possible occurrence of adverse effects after treatment with the plant protection product on transformation processes or on the quality of their products.
|
|
|
Circumstances in which required
|
|
|
When the treated plants or plant products are normally intended for use in transformation process such as wine making, brewing or bread making and when at harvest significant residues are present, the possibility of the occurrence of adverse effects must be investigated and reported where:
|
|
|
— there are indications that the use of the plant protection product could have an influence on the processes involved (for example in the case of use of plant growth regulators or fungicides close to harvest), or
|
|
|
— other products based on the same or a closely similar active substance have been shown to have an adverse influence on these processes or its products.
|
|
|
It is generally sufficient to perform the test with the main formulation type to be authorized.
|
|
6.4.3
|
Effects on the yield of treated plants or plant products
|
|
|
Aim of the tests
|
|
|
The tests shall provide sufficient data to permit an evaluation of the performance of the plant protection product and of the possible occurrence of yield reduction or loss in storage of treated plants or plant products.
|
|
|
Circumstances in which required
|
|
|
The effects of plant protection products on the yield or yield components of treated plants or plant products must be determined where relevant. When treated plants or plant products are likely to be stored the effect on the yield after storage, including data on storage life must be determined where relevant.
|
|
|
This information will normally be available from the tests required under the provisions of point 6.2.
|
|
6.5
|
Phytotoxicity to target plants (including different cultivars), or to target plant products
|
|
|
Aim of the tests
|
|
|
The tests shall provide sufficient data to permit an evaluation of the performance of the plant protection product and of the possible occurrence of phytotoxicity after treatment with the plant protection product.
|
|
|
Circumstances in which required
|
|
|
For herbicides and for other plant protection products for which adverse effects, however transitory, are seen during the trials, performed in accordance to point 6.2, the margins of selectivity on target crops must be established, using twice the recommended rate of application. Where serious phytotoxic effects are seen, an intermediate application rate must also be investigated.
|
|
|
Where adverse effects occur, but are claimed to be unimportant in comparison with the benefits of use or to be transient, evidence to support this claim is required. If necessary yield measurements must be submitted.
|
|
|
The safety of a plant protection product to the main cultivars of the main crops for which it is recommended must be demonstrated, including effects of crop growth stage, vigour, and other factors which may influence susceptibility to damage or injury.
|
|
|
The extent of investigation necessary on other crops will depend on their degree of similarity to the main crops already tested, the quantity and quality of data available on those main crops and how far the manner of use of the plant protection product, if relevant, is similar. It is generally sufficient to perform the test with the main formulation type to be authorized.
|
|
|
Where proposed label claims include recommendations for the use of the plant protection product with other plant protection product(s) or adjuvant(s), the provision of the previous paragraphs apply for the mixture.
|
|
|
Test guideline
|
|
|
Observations concerning phytotoxicity must be recorded and reported in the tests provided for under point 6.2.
|
|
|
Where phytotoxic effects are seen, they must be accurately assessed and recorded in accordance with EPPO Guideline 135, where testing is conducted within the territory of the state. When conducted within the territory of another Member State, the trials must be carried out in accordance with EPPO Guideline 135, or when the Member State concerned so requires, in accordance with guidelines satisfying at least the requirements of EPPO Guideline 135.
|
|
|
A statistical analysis of results amenable to such analysis must be carried out, where necessary the test guideline used must be adapted to enable such analysis.
|
|
6.6
|
Observations on undesirable or unintended side-effects e.g. on beneficial and other non-target organisms, on succeeding crops, other plants or parts of treated plants used for propagating purposes (e.g. seeds, cuttings, runners)
|
|
6.6.1
|
Impact on succeeding crops
|
|
|
Aim of the information required
|
|
|
Sufficient data must be reported to permit an evaluation of possible adverse effects of a treatment with the plant protection product on succeeding crops.
|
|
|
Circumstances in which required
|
|
|
Where data, generated in accordance with section 9, point 9.1, shows that significant residues of the active substance, its metabolites or degradation products, which have or may have biological activity on succeeding crops, remain in soil or in plant materials, such as straw or organic material up to sowing or planting time of possible succeeding crops, observations must be submitted on effects on the normal range of succeeding crops.
|
|
6.6.2
|
Impact on other plants, including adjacent crops
|
|
|
Aim of the information required
|
|
|
Sufficient data must be reported to permit an evaluation of possible adverse effects of a treatment with the plant protection product on other plants, including adjacent crops.
|
|
|
Circumstances in which required
|
|
|
Observations must be submitted on adverse effects on other plants, including the normal range of adjacent crops, when there are indications that the plant protection product could affect these plants via vapour drift.
|
|
6.6.3
|
Impact on treated plants or plant products to be used for propagation
|
|
|
Aim of the information required
|
|
|
Sufficient data must be reported to permit an evaluation of possible adverse effects of a treatment with the plant protection product on plants or plant products to be used for propagation.
|
|
|
Circumstances in which required
|
|
|
Observations must be submitted on the impact of plant protection products on plant parts used for propagation except where the proposed uses preclude use on crops intended for production of seeds, cuttings, runners or tubers for planting, as appropriate:
|
|
|
(i) For seeds — viability, germination and vigour
|
|
|
(ii) Cuttings — rooting and growth rates
|
|
|
(iii) Runners — establishment and growth rates
|
|
|
(iv) Tubers — sprouting and normal growth
|
|
|
Test guideline
|
|
|
For seeds testing shall be done according to ISTA methods25.
|
|
6.6.4
|
Effects on beneficial and other non-target organisms
|
|
|
Any effects, positive or negative, on the incidence of other harmful organisms, observed in the tests performed in accordance with the requirements of this section, shall be reported. Any observed environmental effects must also be reported, especially effects on wildlife and/or beneficial organisms.
|
|
6.7
|
Summary and evaluation of data presented under 6.1 to 6.6
|
|
|
A summary of all data and information provided under points 6.1 to 6.6 must be provided, together with a detailed and a critical assessment of the data, with particular reference to the benefits that the plant protection product offers, adverse effects that do or may arise and measures necessary to avoid or minimize adverse effects.
|
|
7
|
Toxicological studies
|
|
7.1
|
Acute toxicity
|
|
7.1.1
|
Oral
|
|
7.1.2
|
Percutaneous
|
|
7.1.3
|
Inhalation
|
|
7.1.4
|
Skin and, where relevant, eye irritation
|
|
25 International Rules for Seed Testing, 1985. Proceedings of the International Seed Testing Association, Seed Science and Technology, Volume 13, Number 2,1985.
|
|
7.1.5
|
Skin sensitization
|
|
7.1.6
|
Where appropriate, acute dermal toxicity, skin and eye irritation for combinations of plant protection products for which authorization is sought for use in such combinations
|
|
7.2
|
Operator exposure
|
|
7.2.1
|
Dermal absorption
|
|
7.2.2
|
Likely operator exposure under field conditions, including where relevant quantitative analysis of operator exposure
|
|
7.2.3
|
Available toxicological data relating to non-active substances
|
|
8
|
Residues in or on treated products, food and feed
|
|
8.1
|
Data from supervised trials in crops, food or feeding stuffs, for which authorized use is sought, giving all experimental conditions and details, including residue data concerning the active substance, relevant metabolites and relevant other constituents of the plant protection product, from time of application until harvest, or in the case of post-harvest treatment, breakdown of residues during storage and levels of residues at time of release from storage for marketing. Data should be available for the range of climatic and agronomic conditions likely to be encountered in the proposed area of use.
|
|
8.2
|
Effects of industrial processing and/or household preparation on the nature and magnitude of residues
|
|
8.3
|
Effects on taint, odour, taste or other quality aspects due to residues in or on fresh or processed products
|
|
8.4
|
Estimation of residues in products of animal origin resulting from ingestion of feeding stuffs or resulting from contact with bedding, on the basis of residue data referred to in point 8.1 and studies in livestock referred to in Annex II, Part A, point 6.5
|
|
8.5
|
Residue data in succeeding or rotational crops where presence of residues might be expected
|
|
8.6
|
Proposed pre-harvest intervals for envisaged uses, or withholding periods or storage periods, in the case of post-harvest uses
|
|
8.7
|
Proposed maximum residue levels (MRLs) and justification of the acceptability of these residues
|
|
8.8
|
Summary and evaluation of the residue behaviour on the basis of the data submitted under points 8.1 to 8.7
|
|
9
|
Fate and behaviour in the environment
|
|
|
The information provided must, where relevant, include that referred to in Annex II, part A. point 7, and
|
|
9.1
|
Testing for distribution and dissipation in soil
|
|
9.2
|
Testing for distribution and dissipation in water
|
|
G
|
|
|
9.3
|
Testing for distribution and dissipation in air
|
|
10
|
Ecotoxicological studies
|
|
10.1
|
Effects on birds
|
|
10.1.1
|
Acute oral toxicity
|
|
10.1.2
|
Supervised trials to assess risks to avian species under field conditions
|
|
10.1.3
|
If appropriate, studies on acceptance of bait, granules, or treated seeds by birds
|
|
10.2
|
Effects on aquatic organisms
|
|
10.2.1
|
Acute toxicity to fish
|
|
10.2.2
|
Acute toxicity to Daphnia magna
|
|
10.2.3
|
Overspray study (if toxic to fish or other aquatic organisms and persistent in water) to assess risks to aquatic organisms under field conditions
|
|
10.2.4
|
In case of application in/at surface waters
|
|
10.2.4.1
|
Particular studies with fish and other aquatic organisms
|
|
10.2.4.2
|
Residue data in fish concerning the active substance and including toxicologically relevant metabolites
|
|
10.2.5
|
The studies referred to in Annex II, Part A, points 8.2.2, 8.2.3, 8.2.4, 8.2.6, and 8.2.7 may be required for particular plant protection products
|
|
10.3
|
Effects on other non-target organisms
|
|
10.3.1
|
Effects on terrestrial vertebrates other than birds
|
|
10.3.2
|
Toxicity to honey-bees
|
|
10.3.3
|
Toxicity to foraging bees under field conditions
|
|
10.3.4
|
Effects on beneficial arthropods other than bees
|
|
10.3.5
|
Effects on earthworms and other soil non-target macro-organisms, believed to be at risk
|
|
10.3.6
|
Effects on soil non-target micro-organisms
|
|
10.3.7
|
Available data from biological primary screening in summary form
|
|
11
|
Summary and evaluation of points 9 and 10
|
|
12
|
Further information
|
|
12.1
|
Information on authorization in other countries
|
|
12.2
|
Information on established maximum residue limits (MRL) in other countries
|
|
12.3
|
Proposals including justification for the classification and labelling proposed in accordance with Directive 67/548/EEC and Directive 78/631/EEC
|
|
|
— Hazard symbol(s)
|
|
|
— Indication of danger
|
|
|
— Risk phrases
|
|
|
— Safety phrases
|
|
12.4
|
Proposals for risk and safety phrases in accordance with Article 15(1), (g) and (h) and proposed label
|
|
12.5
|
Specimens of proposed packaging
|
|
| |
PART B
|
| |
Preparations of micro-organisms or viruses
|
| |
(this part does not apply to GMOs where points come under Directive 90/220/EEC)
|
| | |
1
|
Identity of the plant protection product
|
|
1.1
|
Applicant (name, address, etc.)
|
|
1.2
|
Manufacturer of the preparation and the active agent(s) (names, addresses, etc., including location of plants)
|
|
1.3
|
Trade name or proposed trade name and manufacturer's development code number for the plant protection product, if appropriate
|
|
1.4
|
Detailed quantitative and qualitative information on the composition of the plant protection product (active organism(s), insert components, extraneous organisms, etc.)
|
|
1.5
|
Physical state and nature of the plant protection product (emulsifiable concentrate, wettable powder, etc.)
|
|
1.6
|
Use category (insecticide, fungicide, etc.)
|
|
2
|
Technical properties of the plant protection product
|
|
2.1
|
Appearance (colour and odour)
|
|
2.2
|
Storage stability — stability and shelf-life. Effects of temperature, method of packaging and storage, etc. on retention of biological activity
|
|
2.3
|
Methods for establishing storage and shelf-life stability
|
|
2.4
|
Technical characteristics of the preparation
|
|
2.4.1
|
Wettability
|
|
2.4.2
|
Persistent foaming
|
|
2.4.3
|
Suspensibility and suspension stability
|
|
2.4.4
|
Wet sieve test and dry sieve test
|
|
2.4.5
|
Particle size distribution, content of dust/fines, attrition and friability
|
|
2.4.6
|
In the case of granules, sieve test and indications of weight distribution of the granules, at least of the fraction with particle sizes bigger than 1 mm
|
|
2.4.7
|
Content of active substance in or on bait particles, granules, or treated seed
|
|
2.4.8
|
Emulsifiability, re-emulsifiability, emulsion stability
|
|
2.4.9
|
Flowability, pourability and dustability
|
|
2.5
|
Physical and chemical compatibility with other products including plant protection products with which its use is to be authorized
|
|
2.6
|
Wetting, adherence and distribution to target plants
|
|
3
|
Data on applications
|
|
3.1
|
Field of use, e.g. field, glasshouse, food or feed storage, home garden
|
|
3.2
|
Details of intended use, e.g. types of harmful organism controlled and/or plants or plant products to be protected
|
|
3.3
|
Application rate
|
|
3.4
|
Where necessary, in the light of the test results, any specific agricultural, plant health and/or environmental conditions under which the product may or may not be used
|
|
3.5
|
Concentration of active substance in material used (e.g. % concentration in the diluted spray)
|
|
3.6
|
Method of application
|
|
3.7
|
Number and timing of application
|
|
3.8
|
Phytopathogenicity
|
|
3.9
|
Proposed instructions for use
|
|
4
|
Further information on the preparation
|
|
4.1
|
Packaging (type, materials, size, etc.), compatibility of the preparation with proposed packaging materials
|
|
4.2
|
Procedures for cleaning applications equipment
|
|
4.3
|
Re-entry periods, necessary waiting periods or other precautions to protect humans and animals
|
|
4.4
|
Recommended methods and precautions concerning handling, storage, transport
|
|
4.5
|
Emergency measures in case of an accident
|
|
4.6
|
Procedures for destruction of decontamination of the plant protection product and its packaging
|
|
5
|
Analytical methods
|
|
5.1
|
Analytical methods for determining the composition of the plant protection product
|
|
5.2
|
Methods for determining residues in or on treated plants or in or on plant products (e.g. biotest)
|
|
5.3
|
Methods used to show microbiological purity of the plant protection product
|
|
5.4
|
Methods used to show the plant protection product to be free from any human and other mammalian pathogens or, if need be, from honey-bee pathogens
|
|
5.5
|
Techniques used to ensure a uniform product and assay methods for its standardization
|
|
6
|
Efficacy data
|
|
6.1
|
Preliminary range-finding tests
|
|
6.2
|
Field experimentation
|
|
6.3
|
Information on the possible occurrence of the development of resistance
|
|
6.4
|
Effects on the quality and where appropriate on the yield of treated plants or effects on the quality of treated plant products
|
|
6.5
|
Phytotoxicity to target plants (including different cultivars), or to target plant products
|
|
6.6
|
Observations concerning undesirable or unintended side-effects, e.g. on beneficial and other non-target organisms, on succeeding crops, other plants or parts of treated plants used for propagation purposes (e.g. seeds, cuttings, runners)
|
|
6.7
|
Summary and evaluation of data presented under points 6.1 to 6.6
|
|
7
|
Toxicity and/or pathogenicity and infectivity studies
|
|
7.1
|
Oral single dose
|
|
7.2
|
Percutaneous single dose
|
|
7.3
|
Inhalation
|
|
7.4
|
Skin and where relevant eye irritation
|
|
7.5
|
Skin sensitization
|
|
7.6
|
Available toxicological data relating to non-active substances
|
|
7.7
|
Operator exposure
|
|
7.7.1
|
Percutaneous absorption
|
|
7.7.2
|
Likely operator exposure under field conditions, including where relevant quantitative analysis of operator exposure
|
|
8
|
Residues in or on treated products, food and feed
|
|
8.1
|
Residue data concerning the active substance including data from supervised trials in crops, food or feeding stuffs for which authorization for use is sought, giving all experimental conditions and details. Data should be available for the range of different climatic and agronomic conditions encountered in the proposed area of use. It is also necessary to identify viable and non-viable residues in treated crops
|
|
8.2
|
Effects of industrial processing and/or household preparation on the nature and magnitude of residues, if appropriate
|
|
8.3
|
Effects on taint, odour, taste or other quality aspects due to residues on or in fresh or processed products, if appropriate
|
|
8.4
|
Residue data in products of animal origin resulting from ingestion of feeding stuffs or contact with bedding, if appropriate
|
|
8.5
|
Residue data in succeeding or rotational crops where presence of residues might be expected
|
|
8.6
|
Proposed pre-harvest intervals for envisaged uses or withholding periods, or storage periods, in the case of post-harvest uses
|
|
8.7
|
Proposed maximum residue levels (MRLs) and the justification of the acceptability of these levels (for toxins), if appropriate
|
|
8.8
|
Summary and evaluation of the residue behaviour on the basis of the data submitted under points 8.1 to 8.7
|
|
9
|
Fate and behaviour in the environment
|
|
9.1
|
In cases where toxins are produced, data as outlined under Part A, point 9 are required, if appropriate
|
|
10
|
Ecotoxicological studies
|
|
10.1
|
Effects on aquatic organisms
|
|
10.1.1
|
Fish
|
|
10.1.2
|
Studies in Daphnia magna and in species closely related to the target organisms
|
|
10.1.3
|
Studies in aquatic micro-organisms
|
|
10.2
|
Effects on beneficial and other non-target organisms
|
|
10.2.1
|
Effects on honey-bees, if appropriate
|
|
10.2.2
|
Effects on other beneficial organisms
|
|
10.2.3
|
Effects on earthworms
|
|
10.2.4
|
Effects on other soil fauna
|
|
10.2.5
|
Effects on other non-target organisms believed to be at risk
|
|
10.2.6
|
Effects on soil microflora
|
|
11
|
Summary and evaluation of points 9 and 10
|
|
12
|
Further information
|
|
12.1
|
Information on authorization in other countries
|
|
12.2
|
Information on established maximum residue limits (MRLs) in other countries
|
|
12.3
|
Proposals including justification for the classification and labelling proposed in accordance with Directives 67/548/EEC and 78/631/EEC
|
|
|
— Hazard symbol(s)
|
|
|
— Indication of danger
|
|
|
— Risk phrases
|
|
|
— Safety phrases
|
|
12.4
|
Proposals for risk and safety phrases in accordance with Article 15(1)(g) and (h) and proposed label
|
|
12.5
|
Specimens of proposed packaging
|
|
| |
Part 3
|
| |
Annex IV
|
| |
(Annex IV to the Directive of 1991)
|
| |
Pro Memoria
|
| |
Part 4
|
| |
Annex V
|
| |
Annex V to the Directive of 1991)
|
| |
Pro Memoria
|
| |
Part 5
|
| |
Annex VI
|
| |
(Annex VI to the Directive of 1991)
|
| |
Pro Memoria
|
| |
Part 6
|
| |
Annex VII
|
| |
(Annex IX to Council Directive 67/548/EEC as amended by the Directive of 1992 and adapted by Commission Directive 91/410/EEC)
|
| |
PROVISIONS RELATING TO CHILD-PROOF FASTENINGS
|
| | |
1.
|
Re-closable packages
|
|
|
Child-proof fastenings used on re-closable packages shall comply with ISO Standard 8317 (1 July 1989 edition) relating to Child-resistant packages — Requirements and methods of testing for re-closable packages adopted by the International Standard Organization (ISO).
|
|
2.
|
Non-re-closable packages
|
|
|
(pro memoria)
|
|
3.
|
Notes
|
|
|
1. Evidence of conformity with the above standard may be certified only by laboratories which conform with European Standards Series EN 45 000.
|
|
|
2. Specific cases
|
|
|
If it seems obvious that packaging is sufficiently safe for children because they cannot get access to the contents without the help of a tool, the test doesn't need to be performed.
|
|
|
In all other cases and when there are sufficient grounds for doubting the security of the closure for a child, the competent authority may ask the person responsible for putting the product on the market to give him a certificate from a test house described below, that either:
|
|
|
— the type of closure is such that it is not necessary to test to the ISO standard referred to above, or
|
|
|
— the closure has been tested and has been found to conform with the standard referred to above.
|
|
| |
Part 7
|
| |
Annex VIII
|
| |
(Annex II to Council Directive 67/548/EEC as amended by the Directive of 1992 and adapted by Commission Directive 93/21/EEC)
|
| |
SYMBOLS AND INDICATIONS OF DANGER FOR DANGEROUS SUBSTANCES AND PREPARATIONS
|
| | |
E
|
|
O
|
|
|

|
Explosive
|
|
Oxidizing
|
|
F
|
|
F+
|
|
|

|
Highly flammable
|
|
Extremely flammable
|
|
F
|
|
T+
|
|
|

|
Toxic
|
|
Very toxic
|
|
C
|
|
Xn
|
|
|
|
Corrosive
|
|
Harmful
|
|
Xi
|
|
N
|
|
|

|
Irritant
|
|
Dangerous for the environment
|
|
| |
|
| |
Part 8
|
| |
Annex IX
|
| |
(Annex III to Council Directive 67/548/EEC as amended by the Directive of 1992 and adapted by Commission Directive 93/21/EEC)
|
| |
NATURE OF SPECIAL RISKS
|
| | |
R1
|
Explosive when dry.
|
|
R2
|
Risk of explosion by shock, friction, fire or other sources of ignition.
|
|
R3
|
Extreme risk of explosion by shock, friction, fire or other sources of ignition.
|
|
R4
|
Forms very sensitive explosive metallic compounds.
|
|
R5
|
Heating may cause an explosion.
|
|
R6
|
Explosive with or without contact with air.
|
|
R7
|
May cause fire.
|
|
R8
|
Contact with combustible material may cause fire.
|
|
R9
|
Explosive when mixed with combustible material.
|
|
R10
|
Flammable.
|
|
R11
|
Highly flammable.
|
|
R12
|
Extremely flammable.
|
|
R14
|
Reacts violently with water.
|
|
R15
|
Contact with water liberates extremely flammable gases.
|
|
R16
|
Explosive when mixed with oxidising substances.
|
|
R17
|
Spontaneously flammable in air.
|
|
R18
|
In use, may form flammable/explosive vapour-air mixture.
|
|
R19
|
May form explosive peroxides.
|
|
R20
|
Harmful by inhalation.
|
|
R21
|
Harmful in contact with skin.
|
|
R22
|
Harmful if swallowed.
|
|
R23
|
Toxic by inhalation.
|
|
R24
|
Toxic in contact with skin.
|
|
R25
|
Toxic if swallowed.
|
|
R26
|
Very toxic by inhalation.
|
|
R27
|
Very toxic in contact with skin.
|
|
R28
|
Very toxic if swallowed.
|
|
R29
|
Contact with water liberates toxic gas.
|
|
R30
|
Can become highly flammable in use.
|
|
R31
|
Contact with acids liberates toxic gas.
|
|
R32
|
Contact with acids liberates very toxic gas.
|
|
R33
|
Danger of cumulative effects.
|
|
R34
|
Causes burns.
|
|
R35
|
Causes severe burns.
|
|
R36
|
Irritating to eyes.
|
|
R37
|
Irritating to respiratory system.
|
|
R38
|
Irritating to skin.
|
|
R39
|
Danger of very serious irreversible effects.
|
|
R40
|
Possible risks of irreversible effects.
|
|
R41
|
Risk of serious damage to eyes.
|
|
R42
|
May cause sensitization by inhalation.
|
|
R43
|
May cause sensitization by skin contact.
|
|
R44
|
Risk of explosion if heated under confinement.
|
|
R45
|
May cause cancer.
|
|
R46
|
May cause heritable genetic damage.
|
|
R48
|
Danger of serious damage to health by prolonged exposure.
|
|
R49
|
May cause cancer by inhalation.
|
|
R50
|
Very toxic to aquatic organisms.
|
|
R51
|
Toxic to aquatic organisms.
|
|
R52
|
Harmful to aquatic organisms.
|
|
R53
|
May cause long-term adverse effects in the aquatic environment.
|
|
R54
|
Toxic to flora.
|
|
R55
|
Toxic to fauna.
|
|
R56
|
Toxic to soil organisms.
|
|
R57
|
Toxic to bees.
|
|
R58
|
May cause long-term adverse effects in the environment.
|
|
R59
|
Dangerous for the ozone layer.
|
|
R60
|
May impair fertility.
|
|
R61
|
May cause harm to the unborn child.
|
|
R62
|
Possible risk of impaired fertility.
|
|
R63
|
Possible risk of harm to the unborn child.
|
|
R64
|
May cause harm to breastfed babies.
|
|
| |
Combination of R-Phrases
|
| | |
R14/15
|
Reacts violently with water, liberating extremely flammable gases.
|
|
R15/29
|
Contact with water liberates toxic, extremely flammable gas.
|
|
R20/21
|
Harmful by inhalation and in contact with skin.
|
|
R20/22
|
Harmful by inhalation and if swallowed.
|
|
R20/21/22
|
Harmful by inhalation, in contact with skin and if swallowed.
|
|
R21/22
|
Harmful in contact with skin and if swallowed.
|
|
R23/24
|
Toxic by inhalation and in contact with skin.
|
|
R23/25
|
Toxic by inhalation and if swallowed.
|
|
R23/24/25
|
Toxic by inhalation, in contact with skin and if swallowed.
|
|
R24/25
|
Toxic in contact with skin and if swallowed.
|
|
R26/27
|
Very toxic by inhalation and in contact with skin.
|
|
R26/28
|
Very toxic by inhalation and if swallowed.
|
|
R26/27/28
|
Very toxic by inhalation, in contact with skin and if swallowed.
|
|
R27/28
|
Very toxic in contact with skin and if swallowed.
|
|
R36/37
|
Irritating to eyes and respiratory system.
|
|
R36/38
|
Irritating to eyes and skin.
|
|
R36/37/38
|
Irritating to eyes, respiratory system and skin.
|
|
R37/38
|
Irritating to respiratory system and skin.
|
|
R39/23
|
Toxic: danger of very serious irreversible effects through inhalation.
|
|
R39/24
|
Toxic: danger of very serious irreversible effects in contact with skin.
|
|
R39/25
|
Toxic: danger of very serious irreversible effects if swallowed.
|
|
R39/23/24
|
Toxic: danger of very serious irreversible effects through inhalation and in contact with skin.
|
|
R39/23/25
|
Toxic: danger of very serious irreversible effects through inhalation and if swallowed.
|
|
R39/24/25
|
Toxic: danger of very serious irreversible effects in contact with skin and if swallowed.
|
|
R39/23/24/25
|
Toxic: danger of very serious irreversible effects through inhalation, in contact with skin and if swallowed.
|
|
R36/26
|
Very toxic: danger of very serious irreversible effects through inhalation.
|
|
R39/27
|
Very toxic: danger of very serious irreversible effects in contact with skin.
|
|
R39/28
|
Very toxic: danger of very serious irreversible effects if swallowed.
|
|
R39/26/27
|
Very toxic: danger of very serious irreversible effects through inhalation and in contact with skin.
|
|
R39/26/28
|
Very toxic: danger of very serious irreversible effects through inhalation and if swallowed.
|
|
R39/27/28
|
Very toxic: danger of very serious irreversible effects in contact with skin and if swallowed.
|
|
R39/26/27/28
|
Very toxic: danger of very serious irreversible effects through inhalation, in contact with skin and if swallowed.
|
|
R40/20
|
Harmful: possible risk of irreversible effects through inhalation.
|
|
R40/21
|
Harmful: possible risk of irreversible effects in contact with skin.
|
|
R40/22
|
Harmful: possible risk of irreversible effects if swallowed.
|
|
R40/20/21
|
Harmful: possible risk of irreversible effects through inhalation and in contact with skin.
|
|
R40/20/22
|
Harmful: possible risk of irreversible effects through inhalation and if swallowed.
|
|
R40/21/22
|
Harmful: possible risk of irreversible effects in contact with skin and if swallowed.
|
|
R40/20/21/22
|
Harmful: possible risk of irreversible effects through inhalation, in contact with skin and if swallowed.
|
|
R42/43
|
May cause sensitization by inhalation and skin contact.
|
|
R48/20
|
Harmful: danger of serious damage to health by prolonged exposure through inhalation.
|
|
R48/21
|
Harmful: danger of serious damage to health by prolonged exposure in contact with skin.
|
|
R48/22
|
Harmful: danger of serious damage to health by prolonged exposure if swallowed.
|
|
R48/20/21
|
Harmful: danger of serious damage to health by prolonged exposure through inhalation and in contact with skin.
|
|
R48/20/22
|
Harmful: danger of serious damage to health by prolonged exposure through inhalation and if swallowed.
|
|
R48/21/22
|
Harmful: danger of serious damage to health by prolonged exposure in contact with skin and if swallowed.
|
|
R48/20/21/22
|
Harmful: danger of serious damage to health by prolonged exposure through inhalation, in contact with skin and if swallowed.
|
|
R48/23
|
Toxic: danger of serious damage to health by prolonged exposure through inhalation.
|
|
R48/24
|
Toxic: danger of serious damage to health by prolonged exposure in contact with skin.
|
|
R48/25
|
Toxic: danger of serious damage to health by prolonged exposure if swallowed.
|
|
R48/23/24
|
Toxic: danger of serious damage to health by prolonged exposure through inhalation and in contact with skin.
|
|
R48/23/25
|
Toxic: danger of serious damage to health by prolonged exposure through inhalation and if swallowed.
|
|
R48/24/25
|
Toxic: danger of serious damage to health by prolonged exposure in contact with skin and if swallowed.
|
|
R48/23/24/25
|
Toxic: danger of serious damage to health by prolonged exposure through inhalation, in contact with skin and if swallowed.
|
|
R50/53
|
Very toxic to aquatic organisms, may cause long-term adverse effects in the aquatic environment.
|
|
R51/53
|
Toxic to aquatic organisms, may cause long-term adverse effects in the aquatic environment.
|
|
R52/53
|
Harmful to aquatic organisms, may cause long-term adverse effects in the aquatic environment.
|
|
| |
Part 9
|
| |
Annex X
|
| |
(Annex V to Council Directive 78/631)
|
| |
SAFETY ADVICE
|
| |
For pesticides classified as very toxic, toxic, harmful, corrosive or irritant, the following safety advice is compulsory:
|
| | |
No. in Annex IV to Directive 67/548/EEC
|
Standard Phrases
|
|
S2
|
Keep out of reach of children.
|
|
S20/21
|
When using do not eat, drink or smoke.
|
|
S13
|
Keep away from food, drink and animal feeding stuffs.
|
|
For harmful pesticides:
|
|
|
S44
|
If you feel unwell, seek medical advice (show the label where possible).
|
|
For very toxic and toxic pesticides:
|
|
|
S45
|
In case of accident or if you feel unwell, seek medical advice immediately (show the label where possible).
|
|
Depending on the particular nature of the risks of the pesticide, the following safety advice must also be given:
|
|
S22
S23
|
Do not breathe dust.
Do not breathe gas/fumes/vapour/ spray (appropriate wording to be proposed by the manufacturer).
|
|
S27
|
Take off immediately all contaminated clothing.
|
|
S36
S37
S42
|
Wear suitable protective clothing.
Wear suitable gloves.
During fumigation/spraying wear suitable respiratory equipment (appropriate wording to be proposed by the manufacturer).
|
|
When a pesticide is classified as corrosive, the following safety advice must also be given:
|
|
S28
S37
S39
|
After contact with skin, wash immediately with plenty of.......... (to be proposed by the manufacturer).
Wear suitable gloves.
Wear eye/face protection.
|
|
When a pesticide contains phosphoric acid esters, the following advice must also be given:
|
|
S28
|
After contact with skin, wash immediately with plenty of..........(to be proposed by the manufacturer)
|
|
If two or more phrases are required, they can be combined in accordance with Annex XI.
|
|
| |
Part 10
|
| |
Annex XI
|
| |
(Annex IV to Council Directive 67/548/EEC as amended by the Directive of 1992 and adapted by Commission Directive 93/21/EEC)
|
| |
SAFETY ADVICE
|
| | |
S1
|
Keep locked up.
|
|
S2
|
Keep out of the reach of children.
|
|
S3
|
Keep in a cool place.
|
|
S4
|
Keep away from living quarters
|
|
S5
|
Keep contents under.....(appropriate liquid to be proposed by the manufacturer).
|
|
S6
|
Keep under.... (inert gas to be proposed by the manufacturer).
|
|
S7
|
Keep container tightly closed.
|
|
S8
|
Keep container dry.
|
|
S9
|
Keep container in a well-ventilated place.
|
|
S12
|
Do not keep the container sealed.
|
|
S13
|
Keep away from food, drink and animal feeding stuffs.
|
|
S14
|
Keep away from.... (incompatible materials to be indicated by the manufacturer).
|
|
S15
|
Keep away from heat.
|
|
S16
|
Keep away from sources of ignition — No smoking.
|
|
S17
|
Keep away from combustible material.
|
|
S18
|
Handle and open container with care.
|
|
S20
|
When using do not eat or drink.
|
|
S21
|
When using do not smoke.
|
|
S22
|
Do not breathe dust.
|
|
S23
|
Do not breathe gas/fumes/vapour/spray (appropriate wording to be proposed by the manufacturer).
|
|
S24
|
Avoid contact with skin.
|
|
S25
|
Avoid contact with eyes.
|
|
S26
|
In case of contact with eyes, rinse immediately with plenty of water and seek medical advice.
|
|
S27
|
Take off immediately all contaminated clothing.
|
|
S28
|
After contact with skin, wash immediately with plenty of ... (to be proposed by the manufacturer).
|
|
S29
|
Do not empty into drains.
|
|
S30
|
Never add water to this product.
|
|
S33
|
Take precautionary measures against static discharges.
|
|
S35
|
This material and its container must be disposed of in a safe way.
|
|
S36
|
Wear suitable protective clothing.
|
|
S37
|
Wear suitable gloves.
|
|
S38
|
In case of insufficient ventilation, wear suitable respiratory equipment.
|
|
S39
|
Wear eye/face protection.
|
|
S40
|
To clean the floor and all objects contaminated by this material, use ... (to be proposed by the manufacturer).
|
|
S41
|
In case of fire and/or explosion do not breathe fumes.
|
|
S42
|
During fumigation/spraying wear suitable respiratory equipment (appropriate wording to be proposed by the manufacturer).
|
|
S43
|
In case of fire, use ... (indicate in the space the precise type of fire-fighting equipment. If water increases risk, add — Never use water).
|
|
S45
|
In case of accident or if you feel unwell, seek medical advice immediately (show the label where possible).
|
|
S46
|
If swallowed, seek medical advice immediately and show this container or label.
|
|
S47
|
Keep at temperature not exceeding .....°C (to be proposed by the manufacturer).
|
|
S48
|
Keep wetted with ... (appropriate material to be proposed by the manufacturer).
|
|
S49
|
Keep only in the original container.
|
|
S50
|
Do not mix with ... (to be proposed by the manufacturer).
|
|
S51
|
Use only in well ventilated areas.
|
|
S52
|
Not recommended for interior use on large surface areas.
|
|
S53
|
Avoid exposure — obtain special instructions before use.
|
|
S56
|
Dispose of this material and its container to hazardous or special waste collection point.
|
|
S57
|
Use appropriate containment to avoid environmental contamination.
|
|
S59
|
Refer to manufacturer/supplier for information on recovery/recycling.
|
|
S60
|
This material and its container must be disposed of as hazardous waste.
|
|
S61
|
Avoid release to the environment. Refer to special instructions/Safety data sheets.
|
|
S62
|
If swallowed, do not induce vomiting: seek medical advice immediately and show this container or label.
|
|
| |
|
| |
Combination of S-Phrases
|
| | |
S1/2
|
Keep locked up and out of the reach of children.
|
|
S3/7
|
Keep container tightly closed in a cool place.
|
|
S3/9/14
|
Keep in a cool, well ventilated place away from........ (incompatible materials to be indicated by the manufacturer).
|
|
S3/9/14/49
|
Keep only in the original container in a cool, well ventilated place away from...... (incompatible materials to be indicated by the manufacturer).
|
|
S3/9/49
|
Keep only in the original container in a cool, well ventilated place.
|
|
S3/14
|
Keep in a cool place away from........ (incompatible materials to be indicated by the manufacturer).
|
|
S7/8
|
Keep container tightly closed and dry.
|
|
S7/9
|
Keep container tightly closed and in a well ventilated place.
|
|
S7/47
|
Keep container tightly closed and at temperature not exceeding .......°C (to be proposed by the manufacturer).
|
|
S20/21
|
When using do not eat, drink or smoke.
|
|
S24/25
|
Avoid contact with skin and eyes.
|
|
S29/56
|
Do not empty into drains, dispose of this material and its container to hazardous or special waste collection point.
|
|
S36/37
|
Wear suitable protective clothing and gloves.
|
|
S36/37/39
|
Wear suitable protective clothing, gloves and eye/face protection.
|
|
S36/39
|
Wear suitable protective clothing and eye/face protection.
|
|
S37/39
|
Wear suitable gloves and eye/face protection.
|
|
S47/49
|
Keep only in the original container at temperature not exceeding .......°C (to be proposed by the manufacturer).
|
|
| |
Part 11
|
| |
Annex XII
|
| |
ADDITIONAL SAFETY ADVICE
|
| | |
FS1
|
Store/keep in original container, tightly closed, in a safe place/under lock and key/away from damp/sources of heat.
|
|
FS2
|
Store/keep/apply/away from/install out of reach of pets/birds/ bees/fish/(young) children.
|
|
FS3
|
Store unused sachets in a safe place. Do not store half-used sachets.
|
|
FS4
|
Not to be used on food crops.
|
|
FS5
|
For use only on/crop/foodstuff/surface/situation/for control of/process.
|
|
FS6
|
To be used only by (professional) operators (instructed/or trained/in the use of chemical/product/type of product/and familiar with the precautionary measures to be observed).
|
|
FS7
|
Do not spray/apply directly to livestock/poultry/pets/food/food crops.
|
|
FS8
|
Do not apply to clothing/fabric/bedding.
|
|
FS9
|
Do not apply to those surfaces/on which food/feed is stored, prepared/or/eaten/which children are likely to touch.
|
|
FS10
|
Do not prepare/use/place bait/dust in/domestic kitchens/larders/food cupboards/ where human or animal food or water could become contaminated.
|
|
FS11
|
Do not use in wet weather/strong winds.
|
|
|
FS12
|
Avoid all contact by mouth/with skin/eyes.
|
|
|
FS13
|
Avoid contact during pregnancy.
|
|
FS14
|
Wear respiratory equipment*/protective gloves/synthetic rubber gloves/PVC gloves/ goggles/face shield/sou'wester/ overalls/apron/impervious apron/mackintosh/boots/impervious boots/spray mask/dust mask when handling/diluting/applying/spraying the concentrate/liquid/dust/fumigant/bait/product/treated seed/freshly treated timber/ solution.
|
|
|
* a suitable type of respirator and canister should be specified.
|
|
FS 15
|
Do not breathe dust/mist/smoke/fog. (If necessary for personal comfort, wear a mask.) or
|
|
|
Avoid working in spray mist/smoke/fog.
|
|
FS 16
|
Wash off splashes immediately.
|
|
|
or
|
|
|
Wash splashes/dust/powder/concentrate/any contamination/ from skin and eyes immediately.
|
|
FS 17
|
Wash hands and exposed skin/before eating, drinking or smoking and after work/before meals and after work/after use/ handling
|
|
|
or
|
|
|
Wash hands before meals and after work.
|
|
FS 18
|
Extinguish all naked flames/including pilot lights/ when applying the fumigant/dust/ liquid/product.
|
|
FS 19
|
Do not apply in the presence of/avoid/naked flames, hot surfaces/or/unprotected electrical equipment.
|
|
FS 20
|
Do not work in confined spaces or enter spaces in which high concentrations of vapour are present. Where this precaution cannot be observed distance breathing or self contained breathing apparatus must be worn, and the work should be done by trained operators.
|
|
FS 21
|
Ensure adequate ventilation when handling/applying (in confined spaces).
|
|
FS 22
|
Wash all protective clothing thoroughly after use, especially the insides of gloves.
|
|
|
or
|
|
|
Avoid excessive contamination of overalls/clothing and launder regularly.
|
|
FS 23
|
Do not handle seed unnecessarily.
|
|
|
FS 24
|
Not to be used as food or feed.
|
|
|
FS 25
|
Do not re-use sack for food or feed.
|
|
FS 26
|
Do not allow.....(product)..... to come into contact with food or cooking utensils.
|
|
FS 27
|
Protect/cover food preparing equipment and eating utensils from contamination during application/before spraying.
|
|
FS 28
|
Do not exceed use of one unit/strip per..... cu m (cu f).
|
|
|
FS 29
|
Do not apply more than.......... per (state amount).
|
|
FS 30
|
Do not apply more than.......... times per crop/season/time period.
|
|
FS 31
|
Keep children/pets/animals away from treated areas/baits for.......... hours/days.
|
|
FS 32
|
Remove/cover all/food/food processing equipment/eating utensils/foodstuffs/fish bowls/fish tanks/caged birds/pets/ water storage tanks/before spraying/application/ dusting/treatment.
|
|
FS 33
|
Protect exposed water/feed/milk machinery/milk containers from contamination.
|
|
FS 34
|
Do not prepare/use/lay baits/dust/spray where food/ feed/water could become contaminated.
|
|
FS 35
|
Remove exposed milk/collect eggs before application.
|
|
|
or
|
|
|
Remove/all animals/pets/livestock/feed/exposed water/ milk/ collect eggs before application/spraying.
|
|
FS 36
|
Fumigate only under conditions which allow no leakage of gas to adjacent occupied premises.
|
|
FS 37
|
Do not harvest/crops for human/animal consumption/for at least (..........days/weeks) after last application.
|
|
|
or
|
|
|
Do not pick/gather/food/crops within.......... hours/days/weeks of treatment.
|
|
FS 38
|
For use on the following crops with stated minimum interval between last application and harvesting. Any table prepared should be based on cleared uses for the product in question. This safety information may be combined on the label with that on the efficient use of the product.
|
|
FS 39
|
Keep unprotected persons/livestock/pets out of treated areas for at least.......... (interval)/until walls/surfaces are dry/until smoke has cleared.
|
|
FS 40
|
Dangerous/Harmful to livestock. Keep all livestock/out of treated areas/away from treated water/for at least.......... (interval). Bury or remove spillages.
|
|
|
or
|
|
|
Keep livestock out of treated areas for at least.......... (interval)/if poisonous weeds such as ragwort are present.
|
|
FS 41
|
Ventilate treated areas/rooms/confined spaces thoroughly/ before occupying/after application when gas/smoke has cleared.
|
|
FS 42
|
Prevent access to baits by children, domesticated animals and pets, (particularly cats, dogs and pigs).
|
|
|
or
|
|
|
Keep/apply/suspend only in positions inaccessible to children and pets.
|
|
FS 43
|
Use bait containers clearly marked "Poison" at all/ surface baiting points.
|
|
FS 44
|
Remove all remains of dust/bait/and bait containers/and dead rodents/after/at end of/treatment and burn/bury/ destroy/ dispose of safely.
|
|
|
or
|
|
|
Remove exposed dust thoroughly after use and bury/burn.
|
|
FS 45
|
Do not sow/plant/transplant..... (specify crops).......... for at least.......... (interval).
|
|
FS 46
|
Do not use outdoors.
|
|
FS 47
|
Do not use in occupied dwelling-houses.
|
|
FS 48
|
Do not apply from aircraft/the air.
|
|
FS 49
|
Dangerous/Harmful to game/wild/caged/birds/butterflies/ animals/fish/bees/pets. Bury spillages/Do not apply within reach of domestic animals/where animals may lick/come in contact with freshly treated surfaces.
|
|
|
or
|
|
|
|
FS 50
|
Dangerous/Harmful to bees. Do not apply/dust/spray/at flowering stage/crops in open flower/during bee activity. Keep down flowering weeds.
|
|
FS 51
|
Dangerous/Harmful to fish. Do not contaminate ponds, waterways or ditches with the chemical or used container.
|
|
|
or
|
|
|
Do not contaminate ponds, waterways or ditches with chemical or used container.
|
|
FS 52
|
Prevent any surface run off to/from entering storm/drains.
|
|
|
or
|
|
|
Avoid contamination of watercourses/ground.
|
|
FS 53
|
This material and its container must be disposed of in a safe way.
|
|
|
or
|
|
|
Dispose of used generator/packaging safely.
|
|
FS 54
|
All washable containers should be labelled: Wash out container thoroughly/empty washings into spray tank/and dispose of safely/dispose of as follows:.......... (specify).
|
|
FS 55
|
All non-washable containers should be labelled: Empty/and return used/ container (completely) and dispose of safely/dispose of as follows:.......... (specify )/in a safe way.
|
|
|
or
|
|
|
Return empty container as instructed by supplier.
|
|
FS 56
|
Handle with care and mix only in closed container.
|
|
FS 57
|
Keep off skin/away from eyes.
|
|
FS 58
|
Avoid contaminating food
|
|
FS 59
|
Open containers outdoors. (Protect from contact with moisture and keep away from burning or glowing material.
|
|
FS 60
|
Apply solutions from unbreakable containers carrying a pouring tube or similar device.
|
|
FS 61
|
Air/ventilate animal feed for at least.......... hours following fumigation.
|
|
FS 62
|
Keep animals/birds/out of premises where grain is under fumigation or being aired/ ventilated following fumigation.
|
|
FS 63
|
Remove excess dust and air treated fabrics thoroughly before use.
|
|
FS 64
|
Search for and burn/bury all rodent bodies. Do not place in refuse bins or on rubbish tips.
|
|
FS 65
|
Spray only into the air/onto surfaces.
|
|
FS 66
|
Do not handle treated fabrics until dry and air thoroughly before use.
|
|
FS 67
|
Avoid skin contact with/do not wear/freshly treated clothing.
|
|
FS 68
|
Do not use on beehives/beekeeping equipment.
|
|
FS 69
|
Avoid (direct) contact with plant life/leaves of growing plants (until solvent has evaporated).
|
|
FS 70
|
Dispose of contaminated material safely by a method approved by the Waste Disposal Authority.
|
|
FS 71
|
Sawdust from treated timber should be treated as contaminated waste and disposed of safely.
|
|
FS 72
|
Medical Advice.
|
|
|
Situations will arise where it is either desirable or necessary that medical advice additional to that provided in the context of safety phrases S 26, S 44, S 45, and S 46 be provided on labels. In such cases the following criteria apply:
|
|
|
Further Advice Poisons Information Centre, Beaumont Hospital, Dublin 9. Telephone: 01-379964 and 01-379966
|
|
|
Organophosphorous pesticides
|
|
|
Symptoms
|
These may include sweating, headache, weakness, faintness and giddiness, nausea, stomach pains, vomiting, small pupils, blurred vision, muscle twitching.
|
|
|
First Aid
|
If any of the above symptoms occur, particularly if there is known contamination; stop work; remove contaminated clothing; wash exposed skin and hair; prevent all exertion; and call doctor at once and show him the label.
|
|
|
Guide to Doctor
|
|
|
|
Specific treatment
|
1 Where signs and symptoms are present and as early as treatment possible, inject treatment atropine sulphate 2 mg or pro rata for children and repeat (if necessary) until fully atropinised.
|
|
|
|
2 If available administer pralidoxine 1 g by intra-muscular injection. Repeat after 3-4 hours.
|
|
|
Other measures
|
1 Keep airway clear.
|
|
|
|
2 Watch respiration — intubation with endotracheal tube, or tracheotomy may be necessary in conjunction with artificial ventilation.
|
|
|
|
3 Put patient at complete rest in hospital for 24 hours at least.
|
|
|
|
Confirmation Estimate cholinesterase activity (5 ml blood unhaemolysed, of Diagnosis collected in an anticoagulant).
|
|
|
Dinitro pesticides
|
|
|
Symptoms
|
These may include fatigue, excessive and unusual sweating and thirst, with sleeplessness and loss of weight in protracted cases. In severe cases there may be rapidly increasing anxiety and restlessness with an increase in respiration and heart rate.
|
|
|
First Aid
|
If any of the above symptoms occur, particularly if there is known contamination; stop work; remove contaminated clothing; wash thoroughly; keep person at rest in the coolest available place; sponge skin with cold water and give cold water to drink; and call doctor at once and show him the label.
|
|
|
Guide to Doctor
|
|
|
|
Treatment
|
1 Treat suspected cases of poisoning by vigorous cooling, giving abundant fluids and oxygen for dyspnoea.
|
|
|
|
2 Do not give morphine or barbiturates.
|
|
|
|
3 If swallowed, wash out stomach and give activated charcoal.
|
|
|
Diagnosis
|
Estimate blood DNOC/Dinoseb (DNBP) concentration (5 ml blood collected in an anticoagulant).
|
|
|
Carbamate pesticides
|
|
|
Symptoms
|
These include excessive sweating, headache, weakness, faintness and giddiness, nausea, stomach pains, vomiting, small pupils, blurred vision, muscle twitching.
|
|
|
First Aid
|
If any of the above symptoms occur, particularly if there is known contamination; stop work; remove contaminated clothing; wash exposed skin and hair; prevent all exertion; call doctor at once and show him the label.
|
|
|
Guide to Doctor
|
|
|
|
Specific
|
1 Where signs and symptoms are present and as early as possible inject atropine sulphate 3 mg or pro rata for children and repeat if necessary until fully atropinised.
|
|
|
|
2 Do not use pralidoxine.
|
|
|
Other measures
|
1 Keep airway clear.
|
|
|
|
2 Watch respiration — intubation with endotracheal tube or tracheotomy may be necessary in conjunction with artificial ventilation.
|
|
|
|
3 Put patient at complete rest in hospital for 24 hours at least.
|
|
|
Bipyridyl pesticides
|
|
|
Symptoms
|
Following ingestion, nausea, vomiting, abdominal pain and diarrhoea (often bloody) may occur within a few hours and result in severe fluid and electrolyte disturbance. In severe cases, circulatory collapse and coma may occur. The concentrate may cause irritation to skin and eyes.
|
|
|
First Aid
|
Wash concentrate or spray from skin immediately; wash eye splashes with water for 10 to 15 minutes and seek medical attention; if swallowed, induce vomiting, if not already occurring and take patient to hospital immediately.
|
|
|
Guide to Doctor
|
|
|
|
Treatment
|
1 Give stomach washout and at the same time test urine and gastric aspirate for the presence of paraquat or diquat.
|
|
|
|
2 If the test is positive, purge the gastrointestinal tract immediately with up to one litre of a 15% suspension of Fuller's Earth and 200 ml of 20% mannitol in water.
|
|
|
|
3 Give a sodium or magnesium sulphate purgative separately.
|
|
|
|
4 Contact the Poisons Information Centre for further advice on treatment.
|
|
|
Quick Qualitative Test
|
Paraquat and diquat can be detected by reduction to blue or green radical ion with sodium dithionite under alkaline conditions.
|
|
|
|
Quick test capsules can be prepared by mixing the following materials:
|
|
|
|
Sodium dithionite (hydrosulphite)
pH buffer powder
Sodium bicarbonate
|
10g
6g
25g
|
|
|
|
The reagents, thoroughly mixed, and packed in l g amounts in gelatin capsules (gauge 0) can be stored at room temperature in a screw-capped container for at least six months.
|
|
|
|
The test is performed by breaking open the capsule and tipping the contents into 10 ml of urine, and shaking gently until dissolved. A green or blue colour indicates the presence of paraquat or diquat.
|
|
| |
Regulation 5 (3)
|
| |
SECOND SCHEDULE
|
| |

|
| |

|
| |
|
| |
|
| |
|
| |

|
| |

|
| |
|
| |

|
| |

|
| |

|
| |

|
| |
|
| |

|
| |

|
| |

|
| |

|
| |
Regulation 6 (2)
|
| |
THIRD SCHEDULE
|
| |
Good Plant Protection Practice
|
| |
INTRODUCTION
|
| |
The principles of Good Plant Protection Practice (GPPP) provide the basis for the identification of optimal practice in the use of plant protection products. GPPP includes principles relating to the use of individual products in the context of overall plant protection programmes. It provides a practical standard for assessing individual practices, with efficacy, human health, animal health and environmental safety being the principal endpoints.
|
| |
In the context of the provisions of Regulation 6, the conditions of authorization and the conditions of use reflected on approved labels, the principles of Good Plant Protection Practice, define the uses and manner of use which are permitted (see Regulation 6).
|
| |
Within the limits established in the context of the uses for which individual plant protection products are authorized and the conditions and restrictions associated with each such authorization, the principles of good plant protection practice provide the basis for:
|
| |
(i) the choice of active substance and formulation;
|
| |
(ii) the choice of—
|
| |
— dosage (and if appropriate volume),
|
| |
— the number of applications to be used,
|
| |
— their timing,
|
| |
— the application equipment to be used and the method of application,
|
| |
in the context of—
|
| |
— crop factors (e.g. cultivar, sowing rate, timing of sowing, fertilization regime, training system, age, spacing),
|
| |
— climatic and edaphic factors (e.g. topography, soil type, rainfall, temperature, light).
|
| |
— possibilities for cultural and biological control,
|
| |
— cost effectiveness,
|
| |
— the harmful organism spectrum to be controlled,
|
| |
— compatibility between products and identified side-effects;
|
| |
providing an overall and rational schedule for treatment with plant protection products, timed partly by the calendar, partly by crop growth stage/phenology and partly by specific harmful organism warning systems, incorporating as appropriate other means of protection, such that efficacious control of the whole harmful organism spectrum (e.g. pest/disease/weed) is achieved, with the minimum amount of product usage.
|
| |
While Good Plant Protection Practice, permits the use of reduced rates of application and use of products in tank mixes, in certain specified circumstances, it does not permit use of plant protection products for purposes for which the product was not authorized, unless an extension of the field of application of an authorized product has been granted in accordance with Regulation 16 for the use concerned. Good Plant Protection Practice does not permit use at rates of application higher, or frequencies more often, than provided for in the conditions of authorization and the conditions of use reflected on approved labels or than provided for in granting extensions of the field of application of authorized products.
|
| |
Good Plant Protection Practice includes use, where possible, in accordance with the principles of integrated control.
|
| |
GENERAL PRINCIPLES
|
| |
These general principles of GPPP must be read in conjunction with the separate specific principles of GPPP for individual crops and where relevant, harmful organisms, to be issued as guidelines by the competent authority. from time to time.
|
| |
1 Crop factors and cultural control
|
| |
GPPP depends first on good agricultural practice in the everyday sense. Crops should be well managed according to local practice. Measures applied should be cost-effective in relation to the value of the crop. Sowing or planting material should be healthy and general hygiene should be maintained. Resistant or tolerant cultivars should be used if available and the crop should be grown in a way which minimizes the need for product inputs (e.g. rotation sequence, elimination of weeds as potential sources of infection). However, this can only be stated very broadly. Farmers and growers may need to grow a highly susceptible cultivar because of its high quality, or use a certain fertilization regime, sowing rate, pruning system, or other technique, because it favours high yield or the achievement of the required quality of produce.
|
| |
Plant protection practices selected must be safe for the crop to be treated. It is evidently GPPP to avoid products which are phytotoxic to species or cultivars, an aspect that generally is covered by the conditions of authorization for individual plant protection products.
|
| |
2 Conditions of authorization of plant protection products
|
| |
The conditions and restrictions associated with individual authorizations granted, establish limits on the uses for such products. Use other than in accordance with these limits is, by definition, never GPPP. However, it is not GPPP to operate at or near these limits, where, for instance in particular situations, the use of fewer applications, lower rates of application or longer intervals between last application and harvest provide satisfactory plant protection. The aim of GPPP is to ensure use in accordance with a concept of optimum effectiveness. A fundamental principle of GPPP is that all safety advice provided on labels be followed, whether relating to the protection of humans, animals or the environment.
|
| |
3 Local harmful organism spectrum to be controlled and thresholds for action
|
| |
In a given crop, only certain harmful organisms are likely to occur. The spectrum of harmful organisms requiring control varies regionally, and depends on climatic conditions, soil type and other factors. Thus, GPPP is conditioned by control needs. In a given region, various indices can be used to determine whether a given harmful organism will need to be controlled in a given season — population levels at the end of the previous season, threshold levels at the beginning of the season, occurrence of weather and other conditions essential for development of the harmful organism. Therefore, going further than just the timing of applications, GPPP seeks to establish whether a harmful organism needs to be controlled or not.
|
| |
The importance of particular harmful organisms varies from season to season. In general, individual plant protection products are active against a spectrum of pests. It is GPPP to use one plant protection product active simultaneously against two or more harmful organisms to be controlled, if the treatments can be correctly timed, rather than to treat them separately with two or more products. However, against a single harmful organism, a more specific product is to be preferred to a broad-spectrum product. It is necessary to avoid unnecessary use of plant protection products. Thus, carefully adjusted use of specific products, by sparing beneficial organisms or avoiding the appearance of resistance, can reduce the inputs necessary for plant protection. These considerations can become very complex and it is not possible to arrive at a general GPPP principle relating to them.
|
| |
4 Choice of active ingredients and formulations
|
| |
The choice of active ingredients and formulations, for use in particular situations, is constrained by a number of separate elements. There is no general GPPP principle that it is better to use few or many active ingredients, or one type of formulation, rather than another. Each individual formulated product is characterized by its efficacy, cost and side-effects.
|
| |
5 Tank mixing and use of adjuvants
|
| |
It is GPPP to use products in tank mixes (including those with fertilizers) provided that the timing of the application is consistent with GPPP for the products separately, since by reducing the number of spray applications, operator exposure, fuel use, passages through the crop, etc., can be reduced. It is not GPPP to use products which are incompatible in a tank mixture or where their individual efficacy or safety is diminished. This is often specified through the conditions associated with authorizations granted, but is not always so specified. In situations not addressed on product labels, it is GPPP to use products in a tank mix, where on the basis of good experimental evidence relating to the range of conditions arising, generated over at least two growing seasons, their compatibility has been established and through the use of a tank mix economies can be achieved.
|
| |
It is GPPP to use an authorized adjuvant with particular plant protection products, where such use is in accordance with the conditions of the authorizations concerned, or where on the basis of good experimental evidence relating to the range of conditions arising, generated over at least two growing seasons, it has been established that through use of an adjuvant, the effectiveness of the plant protection product is enhanced, or the dosage of the plant protection product may be reduced. It is not GPPP to use an adjuvant with a plant protection product in such a manner that residues of the plant protection product at harvest or following storage are increased. It is not GPPP to use an adjuvant which has not been authorized for use with plant protection products.
|
| |
6 Choice of dosage or volume
|
| |
The maximum dosage permitted is fixed by the conditions associated with authorizations granted. It is not GPPP to use higher doses (as they are not authorized and such use is therefore illegal). A low-dosage treatment may be considered GPPP if there is good experimental evidence relating to the range of conditions arising, generated over at least two growing seasons, to show that it is effective. For tall-growing crops, it is important to apply sprays in the correct volume. Dosage will generally be specified as a concentration in this case, and a treatment will not be GPPP if the volume applied is too high or too low.
|
| |
7 Number, timing and frequency of applications
|
| |
It is GPPP to achieve adequate control by applying only as many treatments as are needed for effective control. This number may vary considerably between seasons or localities. The timing of the first application so that it is neither wastefully too early, nor too late (allowing populations to build up) is a key element in GPPP. Numerous warning systems exist which allow forecasts to be made as to when individual harmful organisms will become active (meteorological, direct monitoring, pheromone traps etc.). In any case, account must be taken of local experience, especially of advisory services and farmers and overall visual observation.
|
| |
It may be possible to continue to use such warning systems to time subsequent applications (against successive generations of an insect, or by detecting infection periods for fungi). It is GPPP to do this as far as is practicable. It should, however, be noted that generations may come to overlap, or overall weather conditions may favour a disease over a long period.
|
| |
There are frequently situations when the only possible GPPP is to treat regularly. It is not GPPP to develop a warning system which is impractically complicated, especially if it does not succeed in reducing the number of applications below those of a reasonable calendar programme. Treating according to a fixed programme of dates, or of phenological stages of the crop, can be GPPP, unless it has clearly been shown that it is possible and practical to use a warning system to reduce the number of applications in most years.
|
| |
Some treatment regimes allow for an interaction of dosage and frequency (higher dose less often, lower dose more often, subject to the limits specified on labels). There is no particular GPPP preference in this respect.
|
| |
The timing of the last application will be determined by what is needed for effective control, subject to the over-riding condition that the pre-harvest interval must be respected. In many cases, it may be GPPP to make the last application long before the pre-harvest interval.
|
| |
8 Equipment and method of application
|
| |
It is GPPP to select equipment and application conditions which ensure that a high proportion of product applied reaches its target, with, for sprays in particular, the minimum wasted as aerial drift or onto the ground. Many factors must be taken into consideration (nozzle type, pressure, spray volume, droplet size, speed, etc.), in selecting the equipment and method of application to be used. However, in making such selections, for each product, care must be taken to ensure that efficacy is maintained. It is especially important that the equipment used be properly calibrated and that the calibration be regularly checked, to ensure that the correct dosage is applied.
|
| |
9 Biological means of control
|
| |
The concept of GPPP relates to plant protection products in general, including formulated micro-biological products and natural enemies which may be introduced into a crop (e.g. Encarsia formosa in glasshouses). GPPP is concerned with the proper use of such products, and with the interaction between chemical products and natural enemies introduced into a crop. GPPP is, however, concerned only up to a certain point with the management of natural enemies which pre-exist in a crop. GPPP must respect the conditions of authorization relevant to use, which seek to protect natural enemies. Integrated control seeks to derive maximum benefit from natural control elements and is therefore evidently GPPP. However, practices which are not consistent with the principles of integrated control can still be GPPP, where it is impractical to apply integrated control practices. If reliance on a biological agent (e.g. typhlodromid mites in orchards) has become a regular component of the control scheme within a crop, then it is GPPP to avoid products which would destroy the agent and thus lead to a need to use more of other products.
|
| |
10 Identified side-effects
|
| |
Side-effects on bees, or on wildlife, are basically covered by the conditions of authorization for individual products, so GPPP will automatically take account of them. Side-effects on natural enemies of harmful organisms, have been considered under "Biological means of control". It is GPPP to seek and consider all up-to-date information on such side-effects.
|
| |
While GPPP requires that label recommendations, designed to minimize impact on the environment be followed, it also requires the judicious selection and use of plant protection products to ensure the avoidance of unacceptable contamination of water (surface and ground, whether or not used for abstraction of drinking water) and to ensure the avoidance of any impact on the long-term abundance and diversity of non-target species.
|
| |
One of the most critical side-effects of product use is to impose a selective pressure for the development of resistant harmful organism populations. It is GPPP to take full account of all reports on appearance of practical resistance and to consider the properties of other active ingredients of the same chemical type. For particular crops, with their harmful organism spectra, recommendations may be made on a resistance avoiding strategy e.g. not to use one class of fungicide against foliar diseases because that would favour resistance in other pathogens later in the season, to use a "sensitive" product not more than once a season, always to use mixed formulations with a multi-site active ingredient. Where such strategies have been defined, it is GPPP to follow them.
|
| |
11. Safety
|
| |
GPPP requires that relevant statutory requirements and official codes of practice for the safety of the operator, consumer and environment, be respected.
|
| |
FOURTH SCHEDULE
|
| |
Regulation 8(1)
|
| |
PART 1
|
| |

|
| |

|
| |

|
| |

|
| |

|
| |
|
| |

|
| |

|
| |

|
| |

|
| |

|
| |
|
| |

|
| |
|
| |

|
| |
PART 2
|
| |
Regulation 8 (2)
|
| |
Guidelines and Criteria for the Preparation and Presentation of Complete Dossiers and of Summary Dossiers for the Inclusion of Active Substances in Annex I and for the Authorization of Plant Protection Products
|
| | |
1
|
INTRODUCTION
|
|
1.1
|
The guidance provided and criteria specified, apply to the presentation and format of complete dossiers and summary dossiers, whether submitted in support of applications for inclusion of active substances in Annex I, the authorization of preparations containing the active substance, the modification of authorizations granted, or in the context of the review or renewal of any such inclusion or authorization.
|
|
1.2
|
While requiring standardization in general layout, subject matter, terminology and units of measurement, applicants nevertheless are required to use expert judgement in preparing the documentation concerned. Within the constraints imposed by the provisions of the Directive, which require the submission of separate Annex II and Annex III dossiers, applicants nevertheless should treat these guidelines as providing a degree of flexibility.
|
|
1.3
|
These guidelines and criteria apply to documentation submitted for consideration, whether submitted by applicants, or by other interested parties wishing to submit technical or scientific information, with regard to the potentially dangerous effects of active substances, plant protection products, or their residues, on human or animal health or the environment, or with regard to the effectiveness of particular preparations for their intended uses.
|
|
1.4
|
The objective is to achieve standardization, to the extent that is practicable and feasible, of the format and presentation of documentation submitted, with a view to:
|
|
|
— ensuring the quality and consistency of the documentation submitted;
|
|
|
— facilitating efficiency and economy in the use of resources necessary for the preparation of that documentation; and
|
|
|
— facilitating efficiency and economy in the use of resources necessary for its evaluation.
|
|
1.5
|
Standard Units, Terms and Abbreviations
|
|
|
—Standard Units
|
— the English language version of Standard International Units must be used in reporting and summarizing tests and studies, although other units, if desired or considered relevant, may be used in parentheses26
|
|
| |
26 Particular attention is drawn to the requirement to use metric units — e.g. in the case of application rates, kg active substance/ha; content of active substance in formulations, g/kg or g/l; content of residues, mg/kg; doses in feeding studies, mg/kg body weight.
|
| | |
|
— Standard Terms and Standard Abbreviations
|
— in the interest of avoiding confusion, standard technical terms and abbreviations as specified in Annex 1 and 2 of this Schedule, must be used — these Annexes will be further developed as required. Where terms and abbreviations not listed are used, a concise explanation of each such term or abbreviation must be provided in the text when it is used for the first time. In addition, a listing of all such terms and abbreviations should be provided as an Annex to each relevant summary document.
|
|
1.6
|
Particular attention is drawn to the requirement for standardisation in the terminology used in the nomenclature and reporting of tumours. In the collection of data and compilation of reports, incidences of benign and malignant tumours must not be combined, unless there is clear evidence of benign tumours becoming malignant with time. Similarly, dissimilar un-associated tumours, whether benign or malignant, occurring in the same organ, must not be combined for reporting purposes. In the interests of avoiding confusion, terminology such as that developed by the American Society of Toxicologic Pathologists 27, or the Hannover Tumour Registry (RENI) should be used in the nomenclature and reporting of tumours. The system used must be identified.
|
|
1.7
|
As well as providing two hard copies of the complete dossier and four hard copies of the summary dossier, applicants should also provide the summary and assessment documentation in electronic form (WordPerfect). Details of the required format to be used with respect to pagination, presentation of tables, diagrams and references are provided in Annex 3 of this Schedule.
|
|
2.
|
DOCUMENTATION REQUIRED
|
|
2.1
|
Introduction
|
|
2.1.1
|
The documentation to be prepared and submitted, should allow a comprehensive understanding of the application and facilitate evaluation and decision making with respect to the criteria specified in Articles 4 and 5 of the Directive of 1991, as appropriate, notwithstanding the clear need for reference by the competent authority, to individual study reports and the detailed data (e.g. data on relevant parameters for individual animals), during the course of evaluating the data concerned.
|
|
2.1.2
|
The documentation required comprises a number of separate elements and should include in the following order:
|
|
| | |
(i) Document A
|
a statement of the context in which the dossier is submitted—
|
|
| |
27 Standardized System of Nomenclature and Diagnostic Criteria — Guides for Toxicologic Pathology.
|
| | |
|
— authorization of a preparation in accordance with Regulation 13, where the provisions relating to comparability (paragraph 2 of Regulation 13) are not invoked,
|
|
|
— authorization of a preparation in accordance with Regulation 13, where the provisions relating to comparability are invoked —paragraph 2 of Regulation 13,
|
|
|
— authorization of a preparation for a provisional period in accordance with Regulation 15,
|
|
|
— authorization of a preparation containing an existing active substance, pending its inclusion in Annex I, in accordance with Regulation 18,
|
|
|
— review of an authorization granted,
|
|
|
— renewal of an authorization granted,
|
|
|
— modification of an authorization granted,
|
|
|
— first inclusion of a new active substance in Annex I,
|
|
|
— first inclusion of an existing active substance in Annex I,
|
|
|
— modification or removal of conditions or restrictions associated with the inclusion of an active substance included in Annex I,
|
|
|
— special review of the inclusion of an active substance in Annex I, where indications exist suggesting that the conditions of inclusion are no longer satisfied, or
|
|
|
— routine review anticipating expiry of the period for which the active substance was included in Annex I (i.e. following expiry of the period of inclusion in Annex I);
|
|
(ii) Document B
|
three copies of existing or proposed labels and where relevant leaflets(see Article 16 (2) of the Directive);
|
|
(iii) Document C
|
where registered or authorized in other EC or OECD countries, copies of relevant authorization registration certificates;
|
|
(iv) Document D
|
where relevant, a copy of each relevant notification submitted to the Commission in the context of Article 4 of Commission Regulation (EEC) No 3600/92;
|
|
(v) Documents E — G
|
unless a dossier in accordance with Annex II is submitted, for every formulant (ingredient other than active substance) included in the preparation, the following -
|
|
Document E
|
— a statement as to whether the substance is permitted in food, animal feeding stuffs, medicines or cosmetics in accordance with Community legislation,
|
|
Document F
|
— a copy of the safety data sheet prepared in accordance with Directive 67/548/EEC, and
|
|
Document G
|
where requested, other toxicological and environmental information and data;
|
|
(vi) Document H
|
details of the uses supported in relation to the proposed inclusion of the active substance in Annex I and/or to the proposed authorization of a plant protection product (GAPs) — the information concerned should be provided using the forms as set out in Annex 4 of this Schedule;
|
|
(vii) Document I-1
|
information and documentation relevant to the provisions of Regulation 10 concerning data protection as set out in Annex 5 of this Schedule;
|
|
(viii) Document I-2
|
where relevant and desired, a statement to indicate the data and information involving industrial and commercial secrets for which confidentiality is requested, in accordance with Regulation 11. Where applicants wish to have data and information involving industrial and commercial secrets treated as confidential, applicants should—
|
|
|
— taking account of the provisions of paragraphs 3 and 4 of Regulation 11 and of Council Directive 90/313/EEC of 7 June 1990 on the freedom of access to information on the environment, provide a listing of the data and information for which confidentiality is requested, clearly cross-referenced, for each item, to the relevant test and study reports, as well as to the dossier summaries and supporting documentation submitted,
|
|
|
— highlight items of information for which confidentiality is requested, in relevant study reports, dossier summaries and supporting documentation, and
|
|
|
— for each item listed, provide a justification for the claim that it is, or constitutes, an industrial and commercial secret;
|
|
(ix) Document J-II and J-III
|
copies of individual test and study reports specified in accordance with Annex II and Annex III (Figure 1)—
|
|
|
— details of the requirements relating to the reporting of individual test and study reports are provided in Annex 6 of this Schedule,
|
|
|
— although Article 5.3 of the Directive of 1991 and Article 6.2 (c) of Commission Regulation No. 3600/92 provide that an Annex III dossier for at least one preparation be submitted, in the case of applications for inclusion of an active substance in Annex I, in order to ensure that any Annex I inclusion embraces all existing and envisaged uses, as appropriate, without unnecessary conditions and restrictions, thereby facilitating authorization of preparations containing the active substance for all such uses, the number of preparations for which an Annex III dossier is submitted should be sufficient to reflect the types of formulations and applications envisaged as well as worst case scenarios for operator, dietary and environmental exposure.
|
|
(x) Documents K— N
|
a summary, evaluation and assessment of each Annex II and Annex III dossier, prepared in accordance with the tiered structure described hereunder, and presented graphically in Figure 1, to include-
|
|
Document K-II
|
— a summary of the individual tests and studies (Tier I), prepared by
|
|
Document K-III
|
— oron behalf of the applicant, together with a list of the documents submitted — see also paragraphs 2.2.1.1 and 2.2.2.1,
|
|
Document L-II
Document L-III
|
a summary and assessment of groups of tests and studies, in the light of relevant evaluative and decision making criteria (Tier II) — see also paragraphs 2.2.1.2 and 2.2.2.2,
|
|
Document M-II
Document M-III
|
a summary and assessment of each dossier, in the light of relevant evaluative and decision making criteria (Tier III) - see also paragraphs 2.2.1.3 and 2.2.2.3,
|
|
Documents L & M
|
— where relevant, in Tier II and Tier III, an evaluation, cross referenced to the supporting documentary evidence, of the relevance of particular studies conducted regionally, to the agricultural, plant health and environmental (including climatic) conditions of other regions, together with the rational for extrapolations proposed, and
|
|
(xi) Document N
|
where application for inclusion of an active substance in Annex I is made, an overall assessment of the application (Tier IV) in the light of relevant evaluative and decision making criteria, the conclusions reached by the applicant on the basis of the dossiers submitted (an Annex II dossier and one or more Annex III dossiers), together with a statement of the proposed conditions and restrictions to be associated with any inclusion of the active substance in Annex I, supported with the rational for the proposals made — see also paragraphs 2.2.3.1 and 2.2.3.2;
|
|
(xii) Document N
|
where application for authorization of a plant protection product is made, an overall assessment of the application (Tier IV) in the light of relevant evaluative and decision making criteria, the conclusions reached by the applicant on the basis of the dossiers submitted (Annex III and where appropriate Annex II dossiers), together with a statement of the proposed conditions and restrictions to be associated with any authorization granted, supported with the rational for the proposals made — see also paragraphs 2.2.3.2 and 2.2.3.3;
|
|
(xiii) Document O
|
where relevant, documentation as to the comparability of the agricultural, plant health and environmental (including climatic) conditions of other regions of the Community to the conditions prevailing in Ireland;
|
|
(xiv) Document P
|
where application for authorization of a plant protection product is made and where relevant in accordance with paragraph 2 of Regulation 13—
|
|
|
— an evaluation, cross referenced to the supporting documentary evidence, as to the comparability of the agricultural, plant health and environmental (including climatic) conditions in the Member State in which authorization has been granted to those occurring in Ireland,
|
|
|
— a statement of the rational used for comparisons made, and
|
|
|
— a statement and assessment of the likely impact of any differences in the agricultural, plant health and environmental (including climatic) conditions arising, and proposals relating to the conditions and restrictions to be associated with any authorization granted, to make any such differences irrelevant;
|
|
(xv)
|
a sample of each active substance, as manufactured, which complies with the specification(s) submitted, together with analytical standards for each component included in the proposed residue definition and, on request, a sample of each inactive isomer and impurity of toxicological or environmental concern present in the active substance as manufactured; and
|
|
(xvi)
|
in the case of applications for authorization of a plant protection product, samples of the proposed packaging
|
|
| | |
2.1.3
|
In preparing the summary documentation and assessments required, applicants should also refer to the criteria for evaluation and decision making with respect to the inclusion of active substances in Annex I and the evaluative and decision making criteria specified in Annex VI. In the case of applications involving a proposal for the inclusion of an active substance in Annex I, to vary the conditions of any such inclusion, or for the renewal of any such inclusion, the applicant's objective should be to produce summaries and assessments which, with little or no amendment, other than the removal of confidential information, could be used as a first draft of the Monograph and draft decision to be prepared by the Rapporteur Member State for consideration by the Standing Committee on Plant Health, in accordance with Article 6 of the Directive of 1991. The same general approach should be followed in the case of applications for the authorization of plant protection products.
|
|
| |
|
| |
Figure 1
|
| |
DOCUMENTATION REQUIRED 1
|
| |
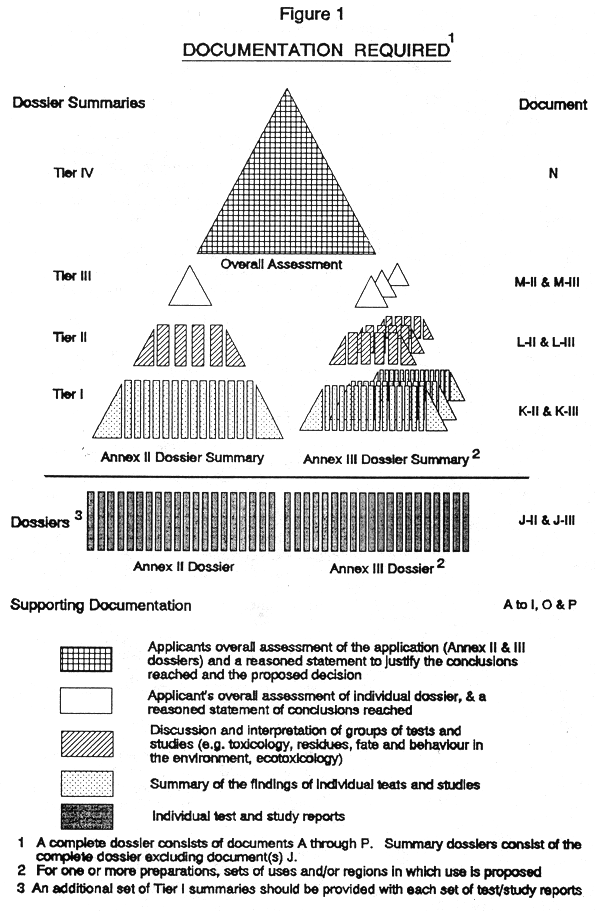
|
| | |
2.2
|
Dossier summary and overall assessments
|
|
2.2.1
|
Annex II dossier
|
|
2.2.1.1
|
Tier I
|
|
(i) Document K-II
|
The dossier summary should address each point of Annex II in the same sequence as in Annex II, contain a summary of each test and study submitted and where relevant, be clearly cross-referenced to the individual test and study reports.
|
|
(ii) Document K-II
|
For each test and study, compiled in the same sequence as in Annex II, the dossier summary should contain in the following order:
|
|
|
— a descriptive title, the test report number, the names of the authors, the date of the report and an indication as to whether it is a published or unpublished report;
|
|
|
— the name and address of the testing facility involved and the dates of commencement and completion of experimental work;
|
|
|
— a statement of the objectives of the study;
|
|
|
— the identity of the test substance and an explicit reference to the relevant specification of composition, and where available, data relevant to the stability and homogeneity of the test substance;
|
|
|
— full details of the composition of any dosing vehicles or solvents used;
|
|
|
— where relevant, information as to the physical form of the test substance or material;
|
|
|
— the identity of the test method and where not a method specified in Annex II, a reasoned justification for the method used and, on request, a copy of the method;
|
|
|
— where test guidelines provide choice as to the method to be used, a reasoned justification for the method used;
|
|
— where deviations from the test guidelines specified, or from other methods used, are employed, a description of and reasoned justification for the deviations;
|
|
— where applicable, an indication that the principles of GLP have been complied with, all divergences and omissions to be identified and justified;
|
|
— where applicable, a clear statement that Good Experimental Practice, Irish/ European Standard IS/EN 45001, or the requirements of points 2.2 and 2.3 of the introduction to Annex III to the Directive of 1991 have been complied with, all divergences and omissions to be identified and justified;
|
|
|
— a description of the test system;
|
|
— a summary of the findings to be reported, as specified in the test Guidelines and in Annex II, clearly referenced to both the individual study reports and the detailed data (e.g. data on relevant parameters for individual animals), where appropriate, in which parameters of relevance to evaluation and decision making are highlighted;
|
|
|
— the identity of any statistical and other techniques applied to the data to aid interpretation, together with adequate documentation thereof where non standard techniques are used;
|
|
— where relevant, an evaluation, cross referenced to the supporting documentary evidence, of the relevance of particular studies conducted in one Member States, to the agricultural, plant health and environmental (including climatic) conditions in other regions of the Community, including Ireland;
|
|
— where reference to published papers is made in reports, the bibliographic references concerned and, on request, copies of the papers concerned; and
|
|
|
— where reference to unpublished data is made (e.g. historical control data on strains of test animals) a summary of such data.
|
|
(iii) Document K-II
|
Where in the case of individual studies, data relating, for instance, to the stability or homogeneity of the test substance is not available (e.g. certain older studies), a justification of the scientific validity of the study should be provided.
|
|
(iv) Document K-II
|
Notwithstanding the general requirement for detailed information relating to GLP for each individual test or study, in certain cases (e.g. physical and chemical properties and further information on the active substance), it is more appropriate that the summaries of the tests and studies concerned be preceded by a general statement as to compliance with GLP. However, in all such cases, each instance of non compliance with or of divergence or omissions from the relevant requirements, should be justified for each individual study.
|
|
(v) Document K-II
|
In the case of information on the identity of the active substance (Annex II, point 1), on the physical and chemical properties of the active substance (Annex II, point 2) and further information on the active substance (Annex II, point 3), a tabular format may be used, taking care to include all essential data (e.g. in the case of physical and chemical properties, the method used, data on the number of observations made, the range of the results obtained, as well as the actual finding to be reported, for each relevant parameter). A suggested format for the presentation of such information is contained in Annex 7 of this Schedule. In other cases a tabular format supported with explanatory text may provide an appropriate means for the presentation of the data and information concerned.
|
|
(vi) Document K-II
|
The final part of the Tier I summary should comprise a listing of all test and study reports, test guidelines, and published papers submitted as part of the dossier. The listing should cover each section separately. For each point or sub-section within sections, the list should be arranged alphabetically by author. For each test and study report, an indication should be provided as to whether it is published or unpublished. The separate sections for which a listing is required are as follows:
|
|
|
1 — Identity of the active substance (Annex II, Point 1),
|
|
— Physical and chemical properties of the active substance (Annex II, Point 2),
|
|
— Further information on the active substance (Annex II, Point 3), and
|
|
— Proposals including justification for the proposals for the classification and labelling of the active substance according to Directive 67/548/EEC (Annex II, Point 10);
|
|
2 — Analytical methods, excluding those for food and feed (Annex II, Points 4.1, 4.2.2, 4.2.3, 4.2.4 and 4.2.5), as well as
|
|
|
— Analytical methods, for food and feed (Annex II, Point 4.2.1);
|
|
3 — Toxicological and metabolism studies on the active substance (Annex II, Point 5);
|
|
4 — Residues in or on treated products, food or feed (Annex II, Point 6);
|
|
5 — Fate and behaviour in the environment (Annex II, Point 7); and
|
|
6 — Ecotoxicological studies on the active ingredient (Annex II, Point 8).
|
|
2.2.1.2
|
Tier II
|
|
(i) Document L-II
|
The second tier summary should contain six sections such that it contains a discussion and interpretation of the results of all Annex II tests and studies and within each section, the conclusions reached. The six sections, which broadly correspond to the main headings of Annex II, are those specified in paragraph 2.2.1.1 (vi).
|
|
(ii) Document L-II
|
Each tier two summary should be confined to and rely only on that data and information contained in the Annex II dossier concerned. If desired, a reference to corresponding Annex III summaries can be included.
|
|
(iii) Document L-II
|
In the case of non submission of particular studies, full justifications should be provided.
|
|
(iv) Document L-II
|
In the case of Section 1 (Identity of the active substance, Physical and chemical properties of the active substance, Further information on the active substance and Proposals including justification for the proposals for the classification and labelling of the active substance according to Directive 67/548/EEC), and those elements of Section 3 (Toxicological and metabolism studies on the active substance) relating to acute toxicity and to mutagenicity, the discussion and interpretation of the data should be supported by a concise tabular presentation of the data.
|
|
(v) Document L-II
|
Where relevant, an evaluation, cross referenced to the supporting documentary evidence, of the relevance of particular studies conducted regionally, to the agricultural, plant health and environmental (including climatic) conditions of other regions, together with the rational for extrapolations proposed, should be included.
|
|
(vi) Document L-II
|
Within each section, it is necessary that each critical point be highlighted, having regard to:
|
|
|
— the extent, quality and consistency of the data;
|
|
— the criteria specified in Article 5 of Directive 91/414/EEC;
|
|
— the criteria and guidelines for evaluation and decision making with respect to the inclusion of active substances in Annex I, where they exist; and
|
|
— the evaluative and decision making criteria specified in Annex VI, to the extent that they are relevant.
|
|
2.2.1.3
|
Tier III
|
|
(i) Document M-II
|
The third tier summary of the Annex II dossier, which should comprise the applicant's overall assessment of the dossier, and a reasoned statement of the conclusions which the applicant believes should be reached on the basis of the data and information provided, should reflect:
|
|
|
— the extent, quality and consistency of the data;
|
|
— the criteria specified in Article 5 of Directive 91/414/EEC;
|
|
— the criteria and guidelines for evaluation and decision making with respect to the inclusion of active substances in Annex I, where they exist; and
|
|
— the evaluative and decision making criteria specified in Annex VI, to the extent that they are relevant.
|
|
(ii) Document M-II
|
Where relevant, an evaluation, cross referenced to the supporting documentary evidence, of the relevance of particular studies conducted regionally, to the agricultural, plant health and environmental (including climatic) conditions of other regions, together with the rational for extrapolations proposed, should be included.
|
|
(iii) Document M-II
|
The assessment and conclusions contained in the third tier summary of the Annex II dossier, should be confined to the data and information provided as part of the Annex II dossier concerned. If desired, a reference to corresponding Annex III summaries can be included.
|
|
2.2.2
|
Annex III dossier
|
|
2.2.2.1
|
Tier I
|
|
(i) Document K-III
|
Each dossier summary should address each point of Annex III in the same sequence as in Annex III, contain a summary of each test and study submitted and where relevant, be clearly cross-referenced to the individual test and study reports.
|
|
(ii) Document K-III
|
Although Article 5.3 of Directive 91/414/EEC and Article 6.2 (c) of Commission Regulation No. 3600/92 provide that an Annex III dossier for at least one preparation be submitted, in the case of applications for inclusion of active substances in Annex I, in order to ensure that the Annex I inclusion embraces all existing and envisaged uses, as appropriate, without unnecessary conditions and restrictions, thereby facilitating authorization of preparations containing the active substance for all such uses, the number of preparations for which an Annex III dossier is submitted should be sufficient to reflect the types of formulations and applications envisaged as well as worst case scenarios for operator, dietary and environmental exposure.
|
|
(iii) Document K-III
|
For each test and study, compiled in the same sequence as in Annex III, each dossier summary should contain in the following order:
|
|
|
— a descriptive title, the test report number, the names of the authors, the date of the report and an indication as to whether it is a published or unpublished report;
|
|
— the name and address of the testing facility involved and the dates of commencement and completion of experimental work;
|
|
— a statement of the objectives of the study;
|
|
— the identity of the test material and an explicit reference to the relevant specification of composition, and where available data relevant to the stability and homogeneity of the test material;
|
|
— full details of the composition of any dosing vehicles or solvents used;
|
|
— where relevant, information as to the physical form of the test material;
|
|
— the identity of the test method and where not a method specified in Annex III, a reasoned justification for the method used and, on request, a copy of the method;
|
|
— where test guidelines provide choice as to the method to be used, a reasoned justification for the method used;
|
|
— where deviations from the test guidelines specified, or from other methods used, are employed, a full description of and reasoned justification for the deviations;
|
|
— where applicable, an indication that the principles of GLP have been complied with, all divergences and omissions to be identified and justified;
|
|
— where applicable, a clear statement that Good Experimental Practice, Irish/European Standard IS/EN 45001, or the requirements of points 2.2 and 2.3 of the introduction to Annex III to the Directive of 1991 have been complied with, all divergences and omissions to be identified and justified;
|
|
— a description of the test system;
|
|
— a summary of the findings to be reported, as specified in the test Guidelines and in Annex III, clearly referenced to both the individual study reports and the detailed data (e.g. data on relevant parameters for individual animals), where appropriate, in which parameters of relevance to evaluation and decision making are highlighted;
|
|
— the identity of any statistical and other techniques applied to the data to aid interpretation, together with adequate documentation thereof where non standard techniques are used;
|
|
— where relevant, an evaluation, cross referenced to the supporting documentary evidence, of the relevance of particular studies conducted in one Member States, to the agricultural, plant health and environmental (including climatic) conditions in other regions of the Community, including Ireland;
|
|
— where reference to published papers is made in reports, the bibliographic references concerned and, on request, copies of the papers concerned; and
|
|
— where reference to unpublished data is made (e.g. historical control data on strains of test animals) a summary of such data.
|
|
(iv) Document K-III
|
Where in the case of individual studies, data relating, for instance, to the stability or homogeneity of the test substance is not available (e.g. certain older studies), a justification of the scientific validity of the study should be provided.
|
|
(v) Document K-III
|
Notwithstanding the general requirement for detailed information relating to GLP for each individual test or study, in certain cases (e.g. physical and chemical properties and further information on the plant protection product, efficacy data, residues in or on treated products, food or feed), it is more appropriate that the summaries of the tests and studies concerned be preceded by a general statement as to compliance with GLP. However, in all such cases, each instance of non compliance with, or of divergence or omissions from the relevant requirements, should be justified for each individual study.
|
|
(vi) Document K-III
|
In the case of information on the identity of the plant protection product (Annex III, point 1), on the physical and chemical properties of the plant protection product (Annex III, point 2), data on application (Annex III, point 3), further information on the plant protection product (Annex III, point 4), efficacy data (Annex II, point 6) and residues in or on treated products, food and feed (Annex III, point 8), a tabular format may be used, taking care to include all essential data (e.g. in the case of physical and chemical properties, the method used, data on the number of observations made, the range of the results obtained, as well as the actual finding to be reported, for each relevant parameter). A suggested format for the presentation of such information is contained in Annex 7 of this Schedule. However, the form as set out in Annex 7 of this Schedule, should be used in reporting data from supervised trials relating to residues in or on treated products, food and feed. In other cases a tabular format supported with explanatory text may provide an appropriate means for the presentation of the data and information concerned.
|
|
(vii) Document K-III
|
The final part of the Tier I summary should comprise a listing of all test and study reports, test guidelines, and published papers submitted as part of the dossier. The listing must cover each section separately. For each point or subsection within sections, the list should be arranged alphabetically by author. For each test and study report, an indication should be provided as to whether it is published or unpublished. The separate sections for which a listing is required are as follows:
|
|
|
1 — Identity of the plant protection product (Annex III, Point 1),
|
|
— Physical, chemical and technical properties of the plant protection product (Annex III, Point 2),
|
|
— Data on application (Annex III, Point 3),
|
|
— Further information on the plant protection product,
|
|
— Information on authorizations in other EC and OECD countries (Annex III, Point 12.1),
|
|
— Proposals including justification for the classification and labelling proposed in accordance Council Directive 67/548/EEC and Directive 78/631/EEC (Annex III, Point 12.3), and
|
|
— Proposals for risk and safety phrases in accordance with Article 16(1) (g) and (h) and proposed label (Annex III, Point 12.4);
|
|
2 — Analytical methods, excluding those for food and feed (Annex III, Point 5.1, 5.2.2, 5.2.3, 5.2.4 and 5.2.5,
|
|
as well as
|
|
— Analytical methods, for food and feed (Annex III, Point 5.2.1);
|
|
3 — Efficacy data (Annex III, Point 6)
|
|
4 — Toxicological studies (Annex III, Point 7);
|
|
5 — Residues in or on treated products, food or feed (Annex III, Point 8);
|
|
6 — Fate and behaviour in the environment (Annex III, Point 9); and
|
|
7 — Ecotoxicological studies (Annex III, Point 10).
|
|
2.2.2.2
|
Tier II
|
|
(i) Document L-III
|
The second tier summary(ies) should contain seven sections such that it contains a discussion and interpretation of the results of all Annex III tests and studies and within each section, the conclusions reached. The seven sections, which broadly correspond to the main headings of Annex III, are those listed in paragraph 2.2.2.1 (vii).
|
|
(ii) Document L-III
|
Tier II summaries, which are to consist of a discussion and interpretation of the results of the tests and studies contained in the Annex III dossier, for the purposes of that discussion and interpretation, necessarily should draw on data and information contained in the relevant Annex II dossier.
|
|
(iii) Document L-III
|
In the case of non submission of particular studies, full justifications should be provided.
|
|
(iv) Document L-III
|
In the case of Section 1 (Identity of the plant protection product, Physical, chemical and technical properties of the plant protection product, Data on application, Further information on the plant protection product, Information on authorizations in other EC and OECD countries, Proposals including justification for the classification and labelling proposed in accordance Council Directive 67/548/EEC and Directive 78/631/EEC, and Proposals for risk and safety phrases in accordance with Article 16(1) (g) and (h) and proposed label), Section 3 (Efficacy data), those elements of Section 4 (Toxicological studies) relating to acute toxicity, and Section 5 (Residues in or on treated products, food or feed) the discussion and interpretation of the data should be supported by a concise tabular presentation of the data.
|
|
(v) Document L-III
|
Where relevant, an evaluation, cross referenced to the supporting documentary evidence, of the relevance of particular studies conducted regionally, to the agricultural, plant health and environmental (including climatic) conditions of other regions, together with the rational for extrapolations proposed, should be included.
|
|
(vi) Document L-III
|
Within each section, it is necessary that each critical point be highlighted, having regard to:
|
|
|
— the extent, quality and consistency of the data;
|
|
— the criteria specified in Articles 4 and 5 of Directive 91/414/EEC;
|
|
— the criteria and guidelines for evaluation and decision making with respect to the inclusion of active substances in Annex I, where they exist and are relevant; and
|
|
— the evaluative and decision making criteria specified in Annex VI.
|
|
2.2.2.3
|
Tier III
|
|
(i) Document M-III
|
The third tier summary of the Annex III dossier, which should comprise the applicant's overall assessment of the dossier, and a reasoned statement of the conclusions which the applicant believes should be reached on the basis of the data and information provided, should reflect:
|
|
|
—the extent, quality and consistency of the data;
|
|
—the criteria specified in Article 5 of Directive 91/414/EEC;
|
|
—the criteria and guidelines for evaluation and decision making with respect to the inclusion of active substances in Annex I where they exist and are relevant; and
|
|
—the evaluative and decision making criteria specified in Annex VI.
|
|
(ii) Document M-III
|
Where relevant, an evaluation, cross referenced to the supporting documentary evidence, of the relevance of particular studies conducted regionally, to the agricultural, plant health and environmental (including climatic) conditions of other regions, together with the rational for extrapolations proposed, should be included.
|
|
(iii) Document M-III
|
The assessment and conclusions contained in the third tier summary of the Annex III dossier, for the purposes of that assessment and the reasoned statement of the conclusions reached, necessarily should draw on data and information contained in the relevant Annex II dossier. However, it should be noted that the complete integration of the conclusions to be derived from Annex II and Annex III data is to be addressed in Tier IV.
|
|
2.2.3
|
Overall assessment of Annex II and III dossiers
|
|
Document N
|
Tier IV
|
|
2.2.3.1
|
This, the final evaluation level, should integrate the conclusions of Annex II and Annex III dossiers and establish the rational for the envisaged Annex I entry or authorization, as appropriate. Consequently, in the case of applications relating to the inclusion of active substances in Annex I, the overall assessment of the Annex II and Annex III dossiers submitted, should be prepared with a view to extending the assessment contained in the Tier III summary of the Annex II dossier to incorporate and reflect relevant elements contained in the Tier III summaries of the Annex III dossiers submitted. It should address all critical elements, having regard to each element of the criteria specified in Article 5 of Directive 91/414/EEC, the criteria and guidelines for evaluation and decision making with respect to the inclusion of active substances in Annex I, where they exist, and to the extent relevant, the evaluative and decision making criteria specified in Annex VI. It should include a proposal relating to the conditions and restrictions to be associated with any inclusion of the active substance in Annex I, together with a detailed justification for the proposals made.
|
|
2.2.3.2
|
Where relevant, a brief summary of the evaluation of the relevance of particular studies conducted regionally, to the agricultural, plant health and environmental including climatic) conditions of other regions, together with the rational for extrapolations proposed, should be included.
|
|
2.2.3.3
|
In the case of an application relating to the authorization of a plant protection product, the fourth tier should comprise the applicant's assessment of the application. It should be prepared with a view to extending the assessment contained in the Tier III summary of the Annex III dossier concerned to incorporate and reflect relevant elements contained in the monograph published by the Commission following the inclusion of the active substance in Annex I, and/or, where relevant to reflect relevant elements contained in the Tier III summary of the Annex II dossier concerned, that are relevant to the authorization of the plant protection product. It should address all critical elements, having regard to each element of the criteria specified in Article 4 of the Directive of 1991 and the evaluative and decision making criteria specified in Annex VI. It should include a proposal relating to the conditions and restrictions to be associated with any authorization granted, together with a detailed justification for the proposals made.
|
|
| |
Annex 1
|
| |
Standard Terms and Abbreviations
|
| | |
ACH
|
acetylcholine
|
|
AChE
|
acetylcholinesterase
|
|
ADI
|
acceptable daily intake
|
|
ADP
|
adenosine diphosphate
|
|
AFID
|
alkali flame-ionization detector or detection
|
|
A/G
|
albumin/globulin ratio
|
|
ai
|
active ingredient
|
|
ALD50
|
approximate median lethal dose, 50%
|
|
AOEL
|
acceptable operator exposure level
|
|
AMD
|
automatic multiple development
|
|
approx.
|
approximate
|
|
as
|
active substance
|
|
at. wt.
|
atomic weight
|
|
ATP
|
adenosine triphosphate
|
|
BA
|
Biological Abstracts (Philadelphia)
|
|
BCF
|
bioconcentration factor
|
|
bfa
|
body fluid assay
|
|
BOD
|
biological oxygen demand
|
|
b.p.
|
boiling point
|
|
BUN
|
blood urea nitrogen
|
|
bw
|
body weight
|
|
|
|
|
c
|
centi- (x 10-2)
|
|
°C
|
degree celsius (centigrade)
|
|
CA
|
Chemical Abstracts
|
|
CAC
|
Codex Alimentarius Commission
|
|
CAD
|
computer aided design
|
|
CAS
|
Chemical Abstracts Service
|
|
CCPR
|
Codex Committee on Pesticide Residues
|
|
CDA
|
controlled drop(let) application
|
|
CEC
|
cation exchange capacity
|
|
cf
|
confer, compare to
|
|
ChE
|
cholinesterase
|
|
CIPAC
|
Collaborative International Pesticides Analytical Council Ltd
|
|
cm
|
centimetre
|
|
CNS
|
central nervous system
|
|
CoC
|
code of conduct
|
|
COD
|
chemical oxygen demand
|
|
COREPER
|
Comite des Representants Permanents
|
|
cu
|
cubic
|
|
cv
|
coefficient of variation
|
|
Cv
|
ceiling value
|
|
cyt
|
cytogenetic analysis
|
|
|
|
|
d
|
day
|
|
DL
|
racemic (optical configuration, a mixture of dextro- and laevo-; preceding a chemical name)
|
|
dlt
|
dominant lethal test
|
|
DMSO
|
dimethylsulfoxide
|
|
DNA
|
deoxyribonucleic Acid
|
|
dnd
|
DNA-damage
|
|
dni
|
DNA-inhibition
|
|
dnr
|
DNA-repair
|
|
dns
|
unscheduled DNA-synthesis
|
|
DO
|
dissolved oxygen
|
|
DOC
|
dissolved organic carbon
|
|
DT
|
disappearance time
|
|
DTH
|
delayed-type hypersensitivity
|
|
|
|
|
EC50
|
effective concentration
|
|
ECD
|
electron capture detector
|
|
ECPA
|
European Crop Protection Association
|
|
EHCD
|
Environmental Health Criteria Document
|
|
EINECS
|
European Inventory of Existing Commercial Chemical Substances
|
|
ELISA
|
enzyme linked immunosorbent assay
|
|
EMDI
|
estimated maximum daily intake
|
|
EP
|
end-use product
|
|
EPPO
|
European and Mediterranean Plant Protection Organization
|
|
ERL
|
extraneous residue limit
|
|
|
|
|
F0
|
parental generation
|
|
F1
|
filial generation, first
|
|
F2
|
filial generation, second
|
|
FAO
|
Food and Agriculture Organization of the UN
|
|
FID
|
flame ionization detector
|
|
f.p.
|
freezing point
|
|
FPD
|
flame photometric detector
|
|
FPLC
|
fast protein liquid chromatography
|
|
|
|
|
g
|
gram
|
|
GAP
|
good agricultural practice
|
|
GATT
|
General Agreement on Tariffs and Trade
|
|
GC-EC
|
gas chromatography with electron capture detector
|
|
GC-MS
|
gas chromatography-mass spectrometry
|
|
GC-MSD
|
gas chromatography with mass selective detection
|
|
GEP
|
good experimental practice
|
|
GFP
|
good field practice
|
|
G.I.
|
gastro-intestinal
|
|
GIT
|
gastro-intestinal tract
|
|
GIFAP
|
Groupement International des Associations Nationales de Fabricants de Produits Agrochimiques
|
|
GL
|
guideline level
|
|
GLC
|
gas liquid chromatography
|
|
GLP
|
good laboratory practice
|
|
GPC
|
gel-permeation chromatography
|
|
GPPP
|
good plant protection practice
|
|
|
|
|
h
|
hour(s)
|
|
ha
|
hectare
|
|
Hb
|
haemoglobin
|
|
HCG
|
human chorionic gonadotropin
|
|
hl
|
hectolitre
|
|
hma
|
host-mediated assay
|
|
HPLC
|
high pressure liquid chromatography or high performance liquid chromatography
|
|
HPPLC
|
high pressure planar liquid chromatography
|
|
HPTLC
|
high performance thin layer chromatography
|
|
HRGC
|
high resolution gas chromatography
|
|
Ht
|
haematocrit
|
|
|
|
|
I50
|
inhibitory dose, 50%
|
|
IARC
|
International Agency for Research on Cancer
|
|
IBT
|
Industrial Bio-Test Laboratories
|
|
IC50
|
median immobilization concentration
|
|
i.d.
|
internal diameter
|
|
ID
|
ionization detector
|
|
i.m.
|
intramuscular
|
|
IMO
|
International Maritime Organisation
|
|
inh
|
inhalation
|
|
IOBC
|
International Organisation for Biological Control of Noxious Animals and Plants
|
|
i.p.
|
intraperitoneal
|
|
IPCS
|
International Programme on Chemical Safety
|
|
IPM
|
integrated pest management
|
|
IR
|
infrared
|
|
ISO
|
International Organization for Standardization
|
|
IUPAC
|
International Union of Pure and Applied Chemistry
|
|
i.v.
|
intravenous
|
|
|
|
|
JECFA
|
FAO/WHO Joint Expert Committee on Food Additives
|
|
FJCMP
|
Joint FAO/WHO Food and Animal Feed Contamination Monitoring Programme
|
|
JMPR
|
Joint Meeting of the FAO Panel of Experts on Pesticide Residues in Food and the Environment and the WHO Expert Group on Pesticide Residues (Joint Meeting on Pesticide Residues)
|
|
|
|
|
k
|
kilo
|
|
kg
|
kilogram
|
|
|
|
|
l
|
litre
|
|
LBC
|
loosely bound capacity
|
|
LC
|
lethal concentration
|
|
LC
|
liquid chromatography
|
|
LC50
|
lethal concentration, median
|
|
LCA
|
life cycle analysis
|
|
LCLo
|
lethal concentration low
|
|
LD50
|
lethal dose, median; dosis letalis media
|
|
LDLo
|
lethal dose low
|
|
LOAEL
|
lowest observable adverse effect level
|
|
LOD
|
limit of determination
|
|
LOEC
|
lowest observable effect concentration
|
|
LOEL
|
lowest observable effect level
|
|
LPLC
|
low pressure liquid chromatography
|
|
LSC
|
liquid scintillation counting or counter
|
|
LT
|
lethal threshold
|
|
|
|
|
m
|
metre
|
|
M
|
molar
|
|
MCH
|
mean corpuscular haemoglobin
|
|
MCHC
|
mean corpuscular haemoglobin concentration
|
|
MCV
|
mean corpuscular volume
|
|
μg
|
microgram
|
|
mg
|
milligram
|
|
min
|
minute(s)
|
|
ml
|
millilitre
|
|
MLD
|
minimum lethal dose
|
|
mm
|
millimetre
|
|
mma
|
microsomal mutagenicity test
|
|
mmo
|
mutation in microorganisms
|
|
mnt
|
micronucleus test
|
|
mo
|
month(s)
|
|
m.p.
|
melting point
|
|
MP
|
manufacturing-use product
|
|
mrc
|
gene conversion and mitotic recombination
|
|
MRE
|
maximum residue expected
|
|
MRL
|
maximum residue limit
|
|
msc
|
mutation in mammalian somatic cells
|
|
MSDS
|
material safety data sheet
|
|
MTD
|
maximum tolerated dose
|
|
|
|
|
n
|
normal (defining isomeric configuration)
|
|
NAEL
|
no adverse effect level
|
|
NATO
|
North Atlantic Treaty Organisation
|
|
NCI
|
National Cancer Institute (USA)
|
|
NCTR
|
National Centre for Toxicological Research (USA)
|
|
n.d.r.
|
not dose-related
|
|
NEDI
|
no effect daily intake (mg/kg body wt/day)
|
|
NEL
|
no effect level
|
|
NERL
|
no effect residue level
|
|
NGO
|
non-governmental organization
|
|
NMR
|
nuclear magnetic resonance
|
|
no.
|
number
|
|
NOAEL
|
no observed adverse effect level
|
|
NOEC
|
no observed effect concentration
|
|
NOED
|
no observed effect dose
|
|
NOEL
|
no observed effect level
|
|
NOIS
|
notice of intent to suspend
|
|
NPD
|
nitrogen-phosphorus detector or detection
|
|
nse
|
non standard exposure
|
|
|
|
|
o
|
ortho (indicating position in a chemical name)
|
|
ODP
|
ozone-depleting potential
|
|
OECD
|
Organization for Economic Cooperation and Development
|
|
OP
|
organophosphorus pesticides
|
|
otr
|
oncogenic transformation
|
|
|
|
|
p
|
para (indicating position in a chemical name)
|
|
Pa
|
pascal
|
|
2-PAM
|
2-pralidoxime
|
|
PAN
|
Pesticide Action Network
|
|
PC
|
paper chromatography
|
|
PCV
|
haematocrit (packed corpuscular volume)
|
|
PD
|
position document
|
|
PEC
|
predicted environmental concentration
|
|
PED
|
plasma-emissions-detector
|
|
pH
|
pH-value
|
|
PHI
|
pre-harvest interval
|
|
pic
|
phage inhibition capacity
|
|
PNEC
|
predicted no effect concentration
|
|
p.o.
|
by mouth
|
|
Pow
|
partition coefficient between n-octanol and water
|
|
|
|
|
ppb
|
parts per billion
|
|
ppm
|
parts per million
|
|
ppq
|
parts per quadrillion
|
|
ppt
|
parts per trillion
|
|
PSP
|
phenolsulfophthalein
|
|
PrT
|
prothrombin time
|
|
PRL
|
practical residue limit
|
|
PT
|
prothrombin time
|
|
PTT
|
partial thromboplastin time
|
|
|
|
|
RAC
|
raw agriculture commodity
|
|
RBC
|
red blood cell
|
|
Rf
|
ratio of fronts
|
|
RL50
|
residual lifetime
|
|
RNA
|
ribonucleic acid
|
|
RNN
|
Re-registration Notification Network
|
|
rns
|
rinsed
|
|
RPM
|
reversed phase material
|
|
RRT
|
relative retention time
|
|
|
|
|
s.c.
|
subcutaneous
|
|
SAC
|
strong adsorption capacity
|
|
SAP
|
serum alkaline phosphatase
|
|
SBLC
|
shallow bed liquid chromatography
|
|
sce
|
sister chromatid exchange
|
|
SD
|
standard deviation
|
|
SE
|
standard error
|
|
SEP
|
standard evaluation procedure
|
|
SF
|
safety factor
|
|
SFC
|
supercritical fluid chromatography
|
|
SFE
|
supercritical fluid extraction
|
|
SI
|
Systeme International d'Unites
|
|
SIMS
|
secondary ion mass spectroscopy
|
|
sin
|
sex chromosome loss and nondisjunction
|
|
sit
|
specific locus test
|
|
SITC
|
Standard International Trade Classification
|
|
sp/spp.
|
species (only after a generic name)
|
|
SPE
|
solid phase extraction
|
|
SPF
|
specific pathogen free
|
|
sp gr
|
specific gravity
|
|
spm
|
sperm morphology
|
|
sq
|
square
|
|
SSD
|
sulphur specific detector
|
|
SSMS
|
spark source mass spectrometry
|
|
STEL
|
short term exposure limit
|
|
SVAT
|
soil-vegetation-atmosphere transfer
|
|
|
|
|
t
|
tonne (metric ton)
|
|
TADI
|
temporary acceptable daily intake
|
|
TBC
|
tightly bound capacity
|
|
TCD
|
thermal conductivity detector
|
|
TCLo
|
toxic concentration, low
|
|
TD
|
thermionic detector, alkali flame detector
|
|
TDLo
|
toxic dose low
|
|
tert
|
tertiary (in a chemical name)
|
|
TEP
|
typical end-use product
|
|
TGAI
|
technical grade of the active ingredient
|
|
TLC
|
thin layer chromatography
|
|
Tlm
|
median tolerance limit
|
|
TLV
|
threshold limit value
|
|
TMDI
|
theoretical maximum daily intake
|
|
TMRC
|
theoretical maximum residue contribution
|
|
TMRL
|
temporary maximum residue limit
|
|
TOC
|
total organic carbon
|
|
trn
|
heritable translocation test
|
|
TWA
|
time weighted average
|
|
|
|
|
UDS
|
unscheduled DNA synthesis
|
|
ULV
|
ultra low volume
|
|
UN
|
United Nations
|
|
UNEP
|
United Nations Environment Programme
|
|
UV
|
ultraviolet
|
|
|
|
|
v/v
|
volume ratio (volume per volume)
|
|
|
|
|
WBC
|
white blood cell
|
|
WHO
|
World Health Organization
|
|
WWF
|
World Wildlife Fund
|
|
wk
|
week
|
|
wt
|
weight
|
|
wt/vol
|
weight per volume
|
|
w/w
|
weight per weight
|
|
|
|
|
yr
|
year
|
|
|
|
|
<
|
less than
|
|
≤
|
less than or equal to
|
|
>
|
greater than
|
|
≥
|
greater than or equal to
|
|
| |
Annex 2
|
| |
Preparation (Formulation) Types and Codes*
|
| | |
|
Code Description
|
Definition
|
|
BR
|
Briquette
|
Solid block designed for controlled releases of active ingredient into water.
|
|
DC
|
Dispersible concentrate
|
A liquid homogeneous preparation to be applied as a solid dispersion after dilution in water.
|
|
EC
|
Emulsifiable concentrate
|
A liquid, homogenous preparation to be applied as an emulsion after dilution in water.
|
|
EO
|
Emulsion, water in oil
|
A fluid, heterogeneous preparation consisting of a dispersion of fine globules of pesticide in water in a continuous organic liquid phase.
|
|
EW
|
Emulsion, oil in water
|
A fluid, heterogeneous preparation consisting of a dispersion of fine globules of pesticide in organic liquid in a continuous water phase.
|
|
PC
|
Gel or paste concentrate
|
A solid preparation to be applied as a gel or a paste after dilution with water.
|
|
SC
|
Suspension concentrate (=flowable concentrate)
|
A stable suspension of active substance(s) in a fluid intended for dilution with water before use.
|
|
CS
|
Capsule suspension
|
A stable suspension of capsules in a fluid normally intended for dilution with water before use.
|
|
SE
|
Suspo-emulsion
|
A fluid, heterogeneous preparation consisting of a stable dispersion of active substance(s) in the form of solid particles and of fine globules in a continuous water phase.
|
|
SG
|
Water soluble granules
|
A preparation consisting of granules to be applied as a true solution of active substance after dissolution in water but may contain insoluble inert ingredients.
|
|
SL
|
Soluble concentrate
|
A liquid homogenous preparation to be applied as a true solution of the active substance after dilution with water.
|
|
SP
|
Water soluble powder
|
A powder preparation to be applied as a true solution of the active substances after solution in water but which may contain insoluble inert ingredients.
|
|
TB
|
Tablet
|
Solid preparation in the form of small, flat plates for dissolution in water.
|
|
WG
|
Water dispersible
|
A preparation granule consisting of granules to be applied after disintegration and dispersion in water.
|
|
WP
|
Wettable powder
|
A powder preparation to be applied as a suspension after dispersion in water.
|
|
OF
|
Oil miscible flowable (= oil active substances) in a miscible suspension)
|
A stable suspension of concentrate fluid intended for dilution in an organic liquid before use.
|
|
OL
|
Oil miscible liquid
|
A liquid, homogenous preparation to be applied as a homogenous liquid after dilution in an organic liquid.
|
|
OP
|
Oil dispersible powder
|
A powder preparation to be applied as a suspension after dispersion in an organic liquid.
|
|
DP
|
Dustable powder
|
A free-flowing powder suitable for dusting.
|
|
GP
|
Flo-dust
|
Very fine dustable powder for pneumatic application in glasshouses.
|
|
ED
|
Electrochargeable liquid
|
Special liquid preparation for electrostatic (electrodynamic) spraying.
|
|
GR
|
Granule
|
A free-flowing solid preparation of a defined granule size range ready for use.
|
|
CG
|
Encapsulated granule
|
A granule with a protective or release controlling coating.
|
|
FG
|
Fine granule
|
A granule in the particle size range from 300 to 2500 æ µ.
|
|
GG
|
Macrogranule
|
A granule in the particle size range from 2000 to 6000 æ µ.
|
|
MG
|
Microgranule
|
A granule in the particle size range from 100 to 600 æ µ.
|
|
SO
|
Spreading oil
|
A preparation designed to form a surface layer on application to water.
|
|
SU
|
Ultra low volume (ULV) suspension
|
A suspension ready for use through ULV equipment.
|
|
TP
|
Tracking powder
|
A rodenticidal contact preparation in powder form.
|
|
UL
|
Ultra low volume (ULV) liquid
|
A homogenous liquid ready for use through ULV equipment.
|
|
AL
|
Other liquids to be applied undiluted
|
Self defining.
|
|
DS
|
Powder for dry seed treatment
|
A powder for application in the dry state directly to seed.
|
|
ES
|
Emulsion for seed treatment
|
A stable emulsion for application to the seed either directly or after dilution.
|
|
FS
|
Flowable concentrate for seed treatment
|
A stable suspension for application to the seed either directly or after dilution.
|
|
LS
|
Solution for seed treatment
|
A solution for application to the seed either directly or after dilution.
|
|
PS
|
Seed coated with a pesticide
|
Self defining.
|
|
SS
|
Water soluble powder for seed treatment
|
A powder to be dissolved in water before application to the seed.
|
|
WS
|
Water dispersible powder for slurry seed treatment
|
A powder to be dispersed at high concentration in water before application as a slurry to the seed.
|
|
AE
|
Aerosol dispenser
|
A container-held preparation which is dispersed generally by a propellant as fine droplets/ particles upon actuation of a valve.
|
|
CB
|
Bait concentrate
|
A solid or liquid intended for dilution before use as a bait.
|
|
FU
|
Smoke generator
|
A combustible preparation generally solid, which upon ignition releases the active substances in the form of a smoke.
|
|
FD
|
Smoke tin
|
Special form of smoke generator.
|
|
FK
|
Smoke candle
|
A smoke generator in the form of a candle.
|
|
FP
|
Smoke cartridge
|
Special form of smoke generator.
|
|
FR
|
Smoke rodlet
|
Special form of smoke generator.
|
|
FT
|
Smoke tablet
|
Special form of smoke generator.
|
|
FW
|
Smoke pellet
|
Special form of smoke generator.
|
|
GA
|
Gas
|
A gas packed in pressure bottle or pressure tank.
|
|
GE
|
Gas generating product
|
A preparation which generates a gas by chemical reaction.
|
|
GS
|
Grease
|
Very viscous preparation based on oil or fat.
|
|
HN
|
Hot fogging concentrate
|
A preparation suitable for application by fogging equipment either directly or after dilution.
|
|
KN
|
Cold fogging concentrate
|
A preparation suitable for application by cold fogging equipment, either directly or after dilution.
|
|
LA
|
Lacquer
|
A solvent based film- forming preparation.
|
|
PA
|
Paste
|
A water based film forming preparation.
|
|
PR
|
Plant rodlet
|
A small rodlet, usually a few centimetres in length and a few millimetres in diameter containing active substance.
|
|
RB
|
Bait (ready for use)
|
A preparation designed to attract and be eaten by the target species.
|
|
AB
|
Grain bait
|
Special forms of bait.
|
|
BB
|
Block baits
|
Special forms of bait.
|
|
GB
|
Granular bait
|
Special forms of bait.
|
|
PB
|
Plate bait
|
Special forms of bait.
|
|
SB
|
Scrap bait
|
Special forms of bait.
|
|
VP
|
Vapour releasing product
|
A preparation containing one or more volatile ingredients, the vapours of which are released into the air. Evaporation rate normally is controlled by using suitable preparations and/or dispensers.
|
|
XX
|
Others
|
|
|
*based upon the catalogue of Pesticide Formulation types and International Coding Systems, developed by GIFAP in co-operation with the German working group on documentation questions. (Arbeitsgruppe EDV Pflanzenschutz Versuchswesen). GIFAP Technical Monograph No 2. 1989.
|
|
| |
Annex 3
|
| |
Requirements Relating to Pagination, Layout, Tables and References
|
| | |
1
|
General
|
|
1.1
|
Documents should be produced using a standard word-processing programme, preferably WordPerfect.
|
|
1.2
|
Both disk and hard copies versions of documents should be submitted.
|
|
1.3
|
One copy of the summary documentation of should be produced with text on one side of pages only, with line spacing of 1½ — or doubled spacing, to facilitate editing. In all copies, tables can be single spaced.
|
|
1.4
|
All programme codes for pagination, font, page numbering. etc. should be at the start of the document. If possible, such codes should not be repeated in the text unless a temporary change is needed. Codes scattered throughout the body of the text can make editing difficult.
|
|
2
|
Format
|
|
2.1
|
As the published text of the monograph to be prepared with the benefit of the dossier summaries is likely to be printed in a 12 cpi (characters per inch) font, such a font if it is available should be used.
|
|
2.2
|
Left/right margins should be 1 inch (2.5 cm) and top/bottom margins 0.5 inch. Lines should be fully justified, with widow/orphan protection.
|
|
2.3
|
Tabs for general text should be set at half-inch (12.5 mm) intervals. If tabs are needed in tables they should be re-set so that a single tab, not a series of tabs, separates sections.
|
|
2.4
|
A page header should be introduced on the top left of each page of the document to show the title of the document, for example: PHORATE Physical and chemical properties of the active substance, or PHORATE Fate and behaviour in the environment, or PHORATE Residues in or on treated products, food and feed.
|
|
3
|
Page numbering
|
|
|
Page numbering should be set to "Top centre". It will then automatically begin on page 1. Do not set either a specific "New page number" or "No page numbers" on any subsequent page — if done, it will create difficulties when individual documents from different sources are assembled into the complete monograph (e.g. pages will be numbered from 1 to say 203 and then either start again at 1 or some other number or be cancelled altogether.
|
|
4
|
Tables
|
|
4.1
|
Tables should be inserted in their intended positions in the text or thereabouts, not at the end of the summary documentation. Although it is customary to insert tables at the end of articles for publication in journals, different considerations apply to the production of camera-ready copy. It facilitates editing if tables are in their correct places from the outset.
|
|
4.2
|
Where possible, the WordPerfect Tables program should be used as it facilitates editing. That program is particularly suitable for tables which are likely to be changed several times, since lines can be added or deleted easily and quickly without affecting the structure of the table.
|
|
4.3
|
As a general rule, separate items of information should be recorded in separate cells of tables.
|
|
4.4
|
Tables created using WordPerfect normally uses lines to separate individual cells. If it is necessary, or desired that the lines be deleted, they can be deleted, but it makes editing easier if they are retained — a note asking the editor to delete them can be included. It is particularly important that cells not be joined (as distinct from deleting lines separating cells), as this almost invariably causes problems with pagination in a table that extends over more than one page.
|
|
4.5
|
Where either portrait (vertical) or landscape (horizontal) formats prove suitable for particular tables, the portrait format is preferred. In the case of data relating to GAPs or residues, the landscape format has to be used. In some circumstances, wide tables can be accommodated vertically by reducing the typeface to 15-17 cpi or by using "Fine" or "Small" attributes. It is particularly important that standard margins are used on all pages including pages with tables. Tables which occupy the full width of a page can be very difficult to edit.
|
|
4.6
|
The caption of a table should not be included within the table itself as it forces the same format to appear on subsequent pages and thus makes it very difficult for the reader to find the beginning of a long table.
|
|
4.7
|
It is generally better not to construct a table covering several pages as a series of separate single-page tables are easier to follow, even though this usually results in a number of partly-filled pages.
|
|
5
|
Diagrams
|
|
|
These can be hand-drawn or photocopies, but where possible, the WordPerfect facility to accept diagrams drawn with graphics software (e.g. DrawPerfect), should be used.
|
|
6
|
References
|
|
|
References to test or study reports, journals and books should be listed alphabetically within each sub-section, in the form shown in the examples below.
|
|
| |
REFERENCES
|
| | |
Point..........
|
|
Fischer, R and Schulze, E-F. 1983a. The effect of Hoe 02782 O F AT202 (fentin acetate, active ingredient 96.4%) on Salmo gairdneri (Rainbow trout) in a static test. Hoechst Pfl. Fo. Biol. Germany. OEK 83 001E. Unpublished.
|
|
Fischer, R and Schulze, E-F. 1983b. The effect of Hoe 29664 O F AT205 (fentin hydroxide, active ingredient 97.0%) on Salmo gairdneri (Rainbow trout) in a static test. Hoechst Pfl. Fo. Biol. Germany. OEK 83 028E. Unpublished.
|
|
Point...........
|
|
Gildemeister, H, Burkle, W.L and Sochor, H. 1985. Hoe 029664-14-C. Anaerobic soil metabolism study with the fungicide triphenyltin hydroxide (TPTH). Hoechst Analyt. Labor, Germany (B) 221/85. Unpublished.
|
|
Point...........
|
|
MacDougall, D. 1964. Guthion. In: Zweig, G. Analytical Methods for Pesticides, Plant Growth Regulators and Food Additives, Vol. II, Academic Press, New York, London.
|
|
Meagher, W.R. Adams, J.M. Anderson, C.A. and MacDougall, D. 1960. Colorimetric determination of Guthion residues in crops. J.Agric Fd Chem 8, 282-286.
|
|
If the first three references were quoted together in the text the citation should be (Fischer and Schulze, 1983a, Fischer and Schulze, 1983b, Gildemeister et al. 1985).
|
|
| |
Annex 4
|
| |
Form for Use in ReportingGood Agricultural Practice (GAP) Information28
|
| |
28 Equivalent to the corresponding Codex and EC forms, but extended to all relevant uses
|
| | |
Responsible body for reporting (name, address) :
|
Date :
|
|
Pesticide(s) (common name(s)) :
|
Page :
|
|
EEC, CIPAC and CCPR No(s). :
|
Country :
|
|
Trade name(s) :
|
|
|
Main uses e.g. insecticide, fungicide :
|
|
|
| |
Use Pattern
|
| | |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
|
Crop and/or situation
|
F or G
|
Pest or group of pests controlled
|
Formulation
|
Application
|
Application rate per treatment
|
PHI
(days)
(k)
|
|
(a)
|
(b)
|
(c)
|
Type (d-f)
|
Conc. of a.s.(i)
|
method
kind (f-h)
|
growth
stage (i)
|
number
(range)
|
kg a.s./hl
|
water 1/ha
|
kg a.i./ha
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
Remarks:
|
| |
( a ) In case of group of crops the EEC and Codex classifications (both) should be used
|
| |
( b ) Outdoor or field use (F), glasshouse application (G) or indoor application (1)
|
| |
( c ) e.g. biting and suckling insects, soil born insects, foliar fungi, weeds
|
| |
( d ) e.g. wettable powder (WP), emulsifiable concentrate (EC), granule (GR)
|
| |
( e ) EEC codes
|
| |
( f ) All abbreviations used must be explained
|
| |
( g ) Method e.g. high volume spraying, low volume spraying, spreading, dusting, drench
|
| |
( h ) Kind, e.g. overall, broadcast, aerial spraying, row, individual plant, between the plants
|
| |
( i ) g/kg or g/l
|
| |
( j ) Growth stage at last treatment
|
| |
( k ) PHI —Pre-harvest interval
|
| |
( l ) Remarks may include: Extent of use/economic importance/restrictions (e.g. feeding/grazing)/minimal intervals between applications
|
| |
Annex 5
|
| |
Information Requirements regarding Data Protection
|
| |
(Document I-1)
|
| | |
1
|
Where in accordance with Regulation 10, it is claimed that the dossiers, or part of the information contained in them, be protected, the owner of the dossiers and/or the studies concerned must be indicated. Where ownership is shared, all of the joint owners must be identified.
|
|
2
|
Where in accordance with sub-paragraphs (1) (a) and (3) (a) of Regulation 10, agreement to the use of information submitted by other parties is claimed, an original signed and notarized letter, confirming such agreement, and submitted by the owner of the information, must be provided. Each such letter must include the following information:
|
|
|
(i) the identity of those to whom agreement for the use of information submitted has been granted;
|
|
|
(ii) the purposes for which such agreement has been granted (a particular product, a group of products, or all relevant products); and
|
|
|
(iii) the period for which the agreement given is valid.
|
|
3
|
In the case of existing active substances being reviewed for possible inclusion in Annex I, or being reviewed in the context of applications for authorization of preparations in accordance with Regulation 18, where the information qualifies for protection pursuant to Regulation 10, the following must be provided:
|
|
|
(i) for each study referred to in sub-paragraph (1) (c) of Regulation 10, a list of the Member States in which one or more preparations containing the active substance was on the market on 24 July 1993, and the dates on which authorization of the first such preparation was granted by each such Member State — in the case of preparations placed on the market prior to 2 December 1985 in Ireland and prior to 6 October 1986 in the UK, the dates of first placing on the market — and the date of expiry of the period of protection for each Member State;
|
|
|
(ii) for each study referred to in sub-paragraph (1) (d) of Regulation 10, a statement that the study was generated for the purposes of achieving inclusion in Annex I or has not been previously submitted to the competent authorities of any of the Member States for the authorization of a plant protection product;
|
|
|
(iii) for each study referred to in sub-paragraph (3) (b) of Regulation 10, the identity of the first Member State to authorize the preparation, the date of authorization and the date of expiry of the period of protection for the Community; and
|
|
|
(iv) for each study referred to in sub-paragraph (3) (c) of Regulation 10, a list of the Member States in which the preparation was authorized, and the dates on which such authorization was granted by each such Member State — in the case of preparations placed on the market prior to 2 December 1985 in Ireland and prior to 6 October 1986 in the UK, the dates of first placing on the market — and the date of expiry of the period of protection for each Member State.
|
|
| |
Annex 6
|
| |
Requirements Concerning the Reporting of Individual Tests and Studies
|
| |
(Documents J-II and J-III)
|
| | |
1
|
The requirements specified hereunder relating to the reporting of tests and studies, supplement those contained in Annex II and Annex III as well as those contained in the testing guidelines and methods specified therein. Accordingly these particular guidelines and criteria, should be read in conjunction with Annex II and Annex III and with relevant test guidelines and test methods.
|
|
2
|
In the case of tests or studies initiated after the 24 July 1993, the methods used must, where relevant, be those specified in Annex II and Annex III. In other cases (e.g. no method specified, old studies), the methods used must be scientifically valid, comparable and appropriate to the issue or parameter to be investigated — see the introductions to Annex II and Annex III — when the reports submitted, in each case, must include or be accompanied by:
|
|
|
— a reasoned justification for the selection of the methodology used, to include where relevant, a comparison in terms of specificity and sensitivity, with the methodology currently specified for such studies;
|
|
|
— a statement of the extent to which the methodology used has been validated;
|
|
|
— where conducted, the results of ring-testing or other validation exercises; and
|
|
|
— a copy of the test guideline, where methods other than those specified in Annex II and Annex III are used.
|
|
3
|
In all cases, the reports of tests and studies must identify, with precision, the methodologies used. In particular, where the guidelines specified or used provide for choice as to the particular methodology to be used, the basis for the choice made must be fully described. All deviations from the prescribed or selected methodology must be fully described and justified. Deviations which involve the investigation of additional parameters, the use of additional groups of animals, exposure levels and durations of exposure, and which are included to preclude the need for the repetition of testing to meet other regulatory requirements — in the case of testing involving vertebrate species, thereby minimizing the total numbers of animals used for testing — where fully described, will be accepted, except where the validity of the test or study has been compromised through such deviation.
|
|
4
|
In the case of tests and studies initiated since the implementation of the principles of good laboratory practice (GLP) for laboratory tests and studies on the 1 July 1988, and for field studies and field elements of studies on 1 January 1993, all test and study reports to which the GLP regimen applies must be signed and dated by the Study Director. The studies concerned are those relating to properties or safety with respect to human health or the environment. In addition, in the case of all such studies:
|
|
|
— where reports of Principal Scientists from co-operating disciplines are included in test or study reports (e.g. field elements of studies), they must be signed and dated;
|
|
|
— each report must be accompanied by a signed Quality Assurance Statement certifying the dates on which inspections were made and the dates any findings were reported to management and to the Study Director;
|
|
|
— the location where all samples, specimens, raw data and the final report are stored, must be stated; and
|
|
|
— corrections and additions to the report must be in the form of an amendment which clearly specifies the reason or reasons for the corrections or additions and must be signed and dated by the Study Director and by the Principal Scientist from each discipline involved.
|
|
5
|
In the case of all other tests and studies initiated since the coming into effect of these Regulations, test and study reports must include all relevant details specified in the context of the Principles of Good Experimental Practice (GEP) (Sixth Schedule) or Irish/European Standard 45001, as appropriate, where conducted within the territory of the state, and include, in addition to the information to be reported as specified in relevant test guidelines:
|
|
|
— the signature and title or an equivalent marking of person(s) accepting technical responsibility for the test report;
|
|
|
— the date of test report; and
|
|
|
— where relevant, corrections or additions to a test report made after it is issued, in the form of an additional document suitably marked, e.g. "Amendment/ Addendum to test report serial number...(or as otherwise identified)".
|
|
6
|
All test and study reports, whether or not conducted in accordance with the Principles of GLP or GEP, must in addition to the information to be reported and specified in accordance with Annex II and Annex III and in the individual testing methods and guidelines, include the following, in the order indicated:
|
|
|
— a descriptive title, the test report number, the names and titles of the authors, the date of the report and an indication as to whether it is a published or unpublished report;
|
|
|
— a clear and explicit reference to the specific Annex II or Annex III point, or points, addressed;
|
|
|
— the name and address of the testing facility involved and the dates of commencement and completion of experimental work;
|
|
|
— a statement of the objectives of the study;
|
|
|
— the identity of the test substance or material and an explicit reference to the relevant specification of composition, and where available, data relevant to the stability and homogeneity of the test substance or material;
|
|
|
— full details of the composition of any dosing vehicles or solvents used;
|
|
|
— where relevant, information as to the physical form of the test substance or material, and as to the concentration, homogeneity and stability of the test substance, substances or material in vehicles or solvents used;
|
|
|
— the identity of the test method and where not a method specified in Annex II or Annex III, a justification for the method used and a copy of the method;
|
|
|
— where test guidelines provide choice as to the method to be used, a justification for the method used;
|
|
|
— where deviations from the test guidelines specified, or from other methods used, are employed, a full description of and justification for the deviations;
|
|
|
— where applicable, a Quality Assurance Statement to indicate that the principles of GLP have been complied with and documented, all divergences and omissions to be identified and justified;
|
|
|
— where applicable, a clear statement that Good Experimental Practice, Irish/European Standard IS/EN 45001, or the requirements of points 2.2 and 2.3 of the introduction to Annex III to the Directive of 1991 have been complied with, all divergences and omissions to be identified and justified;
|
|
|
— a description of the test system;
|
|
|
— a summary of the findings to be reported, as specified in the test Guidelines and in Annex II and Annex III, clearly referenced to the detailed data (e.g. data on relevant parameters for individual animals), where appropriate, together with an analysis of the results in which parameters of relevance to evaluation and decision making are highlighted;
|
|
|
— details of any statistical and other techniques applied to the data to aid interpretation and, where non-standard techniques are used, adequate documentation thereof;
|
|
|
— a discussion and interpretation of the results; and
|
|
|
— where reference to published papers is made in reports copies of the papers.
|
|
| |
Annex 7
|
| |
Format for the Presentation of Data on Physical, Chemical Properties, Technical Characteristics and Certain Other Information
|
| |
Pro Memoria
|
| |
Annex 8
|
| |
Form for Use in Reporting Residues Data from Supervised Trials in Summary Form29
|
| |
29 Equivalent to the corresponding Codex and EC forms, but extended to all relevant uses
|
| | |
Active ingredient (common name) :
|
Producer of commercial product :
|
|
Crop/crop group :
|
Submission date :
|
|
Responsible body for reporting (name, address) :
|
Page :
|
|
Country :
|
Indoor/Outdoor :
|
|
Content of active substance (g/kg or g/l) :
|
Other active substance in the formulation (common name and content) :
|
|
Formulation (e.g. WP) :
|
Residues calculated as :
|
|
Commercial Product (name) :
|
|
|
| | |
1
|
2
|
3
|
|
4
|
|
5
|
6
|
7
|
8
|
9
|
10
|
|
Report No. Location including Postal Code
|
Commodity /Variety
(a)
|
Date of
a) Sowing or Planting
2) Flowering
3) Harvest
(b)
|
Application rate per treatment
|
Dates of treatment(s) or no of treatment(s) and last date (c)
|
Growth stage at last treatment or date
|
Portion analyzed
(a)
|
Residues (mg/kg)
|
PHI (days)
(d)
|
Remarks: (e)
|
|
Kg a.s./hl)
|
Water (1/ha)
|
kg a.s./ha
|
|
|
|
|
|
|
|
|
|
| |
Remarks: (a) According to EEC and Codex class classifications (both) should be used
|
| |
(b) Only if relevant
|
| |
(c) Year must be indicated
|
| |
(d) Days after last application (Label pre-harvest interval, PHI. underline)
|
| |
(e) Remarks may include: Climatic conditions; Reference to analytical method; Information concerning the metabolities included
|
| |
Note: All entries to be filled as appropriate
|
| |
FIFTH SCHEDULE
|
| |
PART 1
|
| |
Regulation 16(2)
|
| |

|
| |

|
| |

|
| |
|
| |

|
| |

|
| |
|
| |

|
| |
PART 2
|
| |
Regulation 16 (2)
|
| |
DOCUMENTATION AND INFORMATION TO SUPPORT APPLICATIONS FOR AN EXTENSION IN THE FIELD OF APPLICATION OF AN AUTHORIZED PLANT PROTECTION PRODUCT
|
| | |
1.
|
INTRODUCTION
|
|
1.1
|
In principle, the information and data requirements relating to human and animal safety, relating to fate and behaviour in the environment and relating to impact on non-target species, to support applications for an extension in the field of application of an authorized plant protection product, are the same as for the authorization of the product concerned for the additional uses, while the data and information requirements relating to efficacy are much less onerous.
|
|
1.2
|
In practice, since in most cases, much of the data relating to human and animal safety, relating to fate and behaviour in the environment and relating to impact on non-target species, will have been supplied by, or on behalf of, the authorization holder, the data and information required for an extension in the field of use of an authorized product, is much less extensive than for the authorization of the product concerned for the additional uses.
|
|
1.3
|
The information and data required and specified in this Schedule, is limited to that which either will always be required, or which may be required, depending on:
|
|
|
(i) the nature of the proposed use;
|
|
|
(ii) the nature and properties of the plant protection product; and
|
|
|
(iii) the nature and extent of the data and information provided by, or on behalf of the authorization holder.
|
|
1.4
|
Since applicants for extensions in the field of use of an authorized plant protection product will not be aware of the extent and nature of the data provided by, or on behalf of, the authorization holder, they should consult the competent authority before undertaking the work necessary for the generation of the data required to support applications.
|
|
1.5
|
In accordance with Regulation 16, extensions in the field of application of authorized plant protection products can only be granted where the uses concerned are minor in nature. Minor uses can be any use on a crop grown to a very limited extent within the territory of the state, or a minor use on crops grown to extensively, taking account, in the case of food crops, of the dietary significance of the food commodity concerned.
|
|
1.6
|
Although a full programme of trials relating to the performance of plant protection products, in terms of their efficacy for the proposed uses and possible phytotoxicity, is not required, sufficient evidence must be provided to establish that it is in the public interest that the extension in the field of use concerned, be granted. The main requirements, in that regard, are:
|
|
|
(i) to demonstrate that crops, plants or plant products cannot be satisfactorily protected by other available means, at an economic cost; and
|
|
|
(ii) to establish that following use as proposed, the desired effect can be achieved without damage to the crop, plants or plant products treated.
|
|
1.7
|
In some cases, it will be possible to fulfil the requirements on the basis of extrapolation from and comparisons with existing authorized uses, or uses authorized in other Member States for the plant protection product or similar plant protection products. Where on the basis of comparisons and extrapolations, it is not established, to the satisfaction of the competent authority, that the plant protection product concerned will provide satisfactory results when used as proposed, a limited programme of trials, conducted in accordance with the requirements of Annex III and the Seventh Schedule, should be undertaken and reported.
|
|
1.8
|
The consideration of applications for extensions in the field of use of authorized plant protection products, must take account of relevant statutory provisions30 and Community instruments not yet implemented31 relating to pesticide residues in food and feed. It is necessary that for each extension in use granted, existing Maximum Residue Levels (MRLs) be respected or sufficient data be provided to support the establishment, on a provisional basis, of a different Community MRL in accordance with the Directive of 1991.
|
|
1.9
|
Existing Community rules relating to the extrapolation of data from crop to crop may serve to preclude the need for or minimize the extent of the data required. In other cases, the authorization holder may have the necessary data on file, and if requested by the applicant, may be prepared to submit it in support of an application for an extension in the field of use of the product concerned. The detailed rules relating to the extent of the data required (see Annex 1 to this Schedule) and the extent to which data available for one crop can be used as a basis for extrapolation to others, including minor crops, is explained in Part 3 of this Schedule. It must be noted that crops which may be considered minor for the purposes of their performance (efficacy and phytotoxicity), may not be treated as minor crops in the context of potential for residues, since in the establishment of MRLs, the issue is judged on a Community basis.
|
|
30 European Communities (Pesticide Residues) (Fruit and Vegetables) Regulations, 1989 (
S.I. No. 105 of 1989
); European Communities (Pesticide Residues) (Feedingstuffs) Regulations, 1992 (
S.I. No. 40 of 1992
); European Communities (Pesticide Residues) (Cereals) Regulations, 1988 and 1993 (
S.I. No. 216 of 1988
and
S.I. No. 316 of 1993
); European Communities (Pesticide Residues) (Foodstuffs of Animal Origin) Regulations, 1988 and 1993 (
S.I. No. 217 of 1988
and
S.I. No. 317 of 1993
).
31 Council Directive 90/642/EEC of 27 November 1990 on the fixing of maximum levels for pesticide residues in and on certain products of plant origin, including fruit and vegetables, O.J. No. 350/71 14/12/1990; Council Directive 93/58/EEC of the 29 June 1993 amending Annex II to Directive 76/895/EEC relating to the fixing of maximum levels for pesticide residues in and on fruit and vegetables and the Annex to Directive 90/642/EEC relating to the fixing of maximum levels for pesticide residues in and on certain products of plant origin, and providing for the establishment of a first list of maximum levels, O.J. No. 211/1, 23/8/1993.
|
|
1.10
|
In all cases in which applicants consider that residue data may be required, they should consult the competent authority prior to undertaking any programme to generate such data, as data of which the applicant is unaware, which might serve to preclude the need for or reduce the extent of any additional data required, may be available to the competent authority. Where it is necessary to generate data relating to residues in food or feed products, the necessary supervised trials must be conducted and reported in accordance with relevant Community rules and procedures (Annex 1 of this Schedule). Since the data to be generated must relate to all relevant parts, where use is envisaged, of the Northern and Central Europe region (see paragraph 4 of Part B of this Schedule), not just to Ireland, the necessary trials programmes must be designed to take account of the range of factors affecting variability in the region concerned. Accordingly, applicants should co-operate with counterparts in other Member States, to ensure that data relevant to the necessary range of conditions is generated and duplication of effort is avoided.
|
|
2.
|
INFORMATION RELATING TO THE APPLICANT
|
|
2.1
|
Applicant (name and address, telephone and telefax numbers, and name of the appropriate person to contact)
|
|
|
The name and address of the applicant (permanent community address) must be provided as must the name, position, telephone and telefax number of the appropriate person to contact.
|
|
|
Where, in addition, the applicant has an office, agent or representative in the territory of the State, the name and address of the local office, agent or representative should be provided, as should the name, position, telephone and telefax number of the appropriate person to contact.
|
|
2.2
|
Type of applicant (official or scientific body involved in agricultural activities, professional agricultural organization, professional user)
|
|
|
Since applications may only be accepted when submitted by official or scientific bodies involved in agricultural activities, agricultural organizations or professional users, applicants must provide sufficient information to demonstrate that they fall within one or more of those categories of applicant.
|
|
3.
|
INFORMATION RELATING TO THE AUTHORIZED PLANT PROTECTION PRODUCT
|
|
3.1
|
Identity of the plant protection product (as printed on labels)
|
|
3.1.1
|
Trade name of the plant protection product.
|
|
3.1.2
|
Name and address of the authorization holder.
|
|
3.1.3
|
Authorization number of the plant protection product.
|
|
3.1.4
|
Identity and content of each active substance in the plant protection product.
|
|
3.1.5
|
Physical state and nature of the preparation (emulsifiable concentrate, wettable powder, . solution etc.).
|
|
3.2
|
Authorized uses of the plant protection product (as printed on labels)
|
|
3.2.1
|
Use category (herbicide, insecticide, etc.).
|
|
3.2.2
|
Field of use, e.g. field, glasshouse, food or feed storage, home garden.
|
|
3.2.3
|
Details of authorized uses, e.g. types of harmful organisms controlled and/or plants or plantproducts protected.
|
|
3.2.4
|
Application rate(s).
|
|
3.2.5
|
Method of application.
|
|
3.2.6
|
Number and timing of applications and duration of protection.
|
|
3.3
|
Crop Safety Intervals (as printed on labels)
|
|
3.3.1
|
Waiting period between application and sowing or planting the crop to be protected.
|
|
3.3.2
|
Waiting period between last application and sowing or planting succeeding crops.
|
|
3.4
|
Re-entry periods, necessary waiting periods or other precautions to protect man, livestock and the environment (as printed on labels)
|
|
3.4.1
|
Pre-harvest intervals for each crop.
|
|
3.4.2
|
Re-entry periods for livestock to areas to be grazed.
|
|
3.4.3
|
Re-entry periods for humans to crops.
|
|
3.4.4
|
Re-entry periods for humans to buildings or treated spaces.
|
|
3.4.5
|
Waiting period between application and handling treated products.
|
|
3.4.6
|
Minimum storage period in the case of post-harvest treatments.
|
|
3.4.7
|
Withholding period for animal feeding stuffs.
|
|
3.4.8
|
Waiting period between last application and sowing or planting succeeding crops.
|
|
4
|
DATA AND INFORMATION RELATING TO EACH PROPOSED USE
|
|
4.1
|
Data on application
|
|
4.1.1
|
Field of use, e.g. field, glasshouse, food or feed storage, home garden
|
|
|
The field(s) of use, existing and proposed, for preparations containing the active substance must be specified from among the following:
|
|
|
Field use — Agriculture
|
|
|
— Horticulture
|
|
|
— Forestry
|
|
|
— Viticulture
|
|
|
Protected crops
|
|
|
Amenity
|
|
|
Weed control on non-cultivated areas
|
|
|
Home gardening
|
|
|
House plants
|
|
|
Plant products storage practice
|
|
4.1.2
|
Details of proposed use, e.g. types of harmful organisms controlled and/or plants or plant products protected
|
|
|
Details of the intended use must be provided.
|
|
|
Where relevant, effects achieved e.g. sprout suppression, retardation of ripening, reduction in stem length, enhanced fertilization etc. must be reported.
|
|
4.1.3
|
Application rate(s)
|
|
|
For each method of application and each use, the rate of application per unit (ha, m2, m3) treated, in terms of g or kg of both preparation and active substance, must be provided.
|
|
|
Application rates shall normally be expressed in g or kg/ha or in kg/m3 and where appropriate in g or kg/tonne; for protected crops and home gardening use rates shall be expressed in g or kg/100 m2 or g or kg/m3.
|
|
4.1.4
|
Method of application
|
|
|
The method of application proposed must be described fully, indicating the type of equipment to be used, if any, as well as the type and volume of diluent to be used per unit of area or volume.
|
|
4.1.5
|
Number and timing of applications and duration of protection
|
|
|
The maximum number of applications to be used and their timing, must be reported. Where relevant the growth stages of the crop or plants to be protected and the development stages of the harmful organisms, must be indicated. Where possible the interval between applications, in days, must be stated.
|
|
|
The duration of protection afforded both by each application and by the maximum number of applications to be used, must be indicated.
|
|
4.1.6
|
Instructions for use
|
|
|
The proposed instructions for use of the preparation, to be provided to workers involved, must be provided.
|
|
4.2
|
Proposed Crop Safety Intervals
|
|
4.2.1
|
Waiting period between application and sowing or planting the crop to be protected.
|
|
4.2.2
|
Waiting period between last application and sowing or planting succeeding crops.
|
|
4.3
|
Proposed re-entry periods, necessary waiting periods or other precautions to protect man, livestock and the environment
|
|
|
The information provided must follow from and be supported by the data provided for the active substance(s) and that provided under sections 7 and 8.
|
|
|
Where relevant, pre-harvest intervals, re-entry periods or withholding periods necessary to minimize the presence of residues in or on crops, plants and plant products, or in or on treated areas or spaces, with a view to protecting man or livestock, must be specified e.g.
|
|
|
— pre-harvest interval (in days) for each relevant crop;
|
|
|
— re-entry period (in days) for livestock, to areas to be grazed;
|
|
|
— re-entry period (in hours or days) for man to crops, buildings or spaces treated;
|
|
|
— withholding period (in days) for animal feedingstuffs;
|
|
|
— waiting period (in days), between application and handling treated products; or
|
|
|
— waiting period (in days), between last application and sowing or planting succeeding crops.
|
|
4.4
|
Evidence that each proposed use is minor in nature
|
|
4.4.1
|
Minor crops
|
|
4.4.1.1
|
The national area of production (ha) and total annual production (tonnes or plants, as appropriate), must be reported.
|
|
4.4.1.2
|
An estimate of potential production area and total annual production which might be treated, must be provided.
|
|
4.4.1.3
|
In the case of food crops a statement as to whether, or not, consumed by infants and of the dietary importance of the food commodity must be provided for-
|
|
|
(i) persons 8 to 18 years of age; and
|
|
|
(ii) persons over 18 years of age,
|
|
4.4.2
|
Major crops
|
|
4.4.2.1
|
The national area of production (ha) and total annual production (tonnes or plants, as appropriate), must be reported.
|
|
4.4.2.2
|
An estimate of potential production area and total annual production which might be treated, must be provided.
|
|
4.4.2.3
|
In the case of food crops a statement as to whether, or not, consumed by infants and of the dietary importance of the food commodity must be provided for -
|
|
|
(i) persons 8 to 18 years of age; and
|
|
|
(ii) persons over 18 years of age.
|
|
4.5
|
Evidence of each proposed use being in the public interest
|
|
4.5.1
|
Alternative plant protection means available, their effectiveness and cost
|
|
|
Where alternative means to achieve the desired effect exist and could be used, their effectiveness and cost, relative to the proposed means, must be reported. Where available, supporting documentary evidence should be provided.
|
|
4.5.2
|
Estimates of losses in yield, quality and of increased production costs due to the plant protection product not being authorized for each use for which an extension is sought
|
|
|
Details, to include costing, should be provided. Where available, supporting documentary evidence should be provided.
|
|
4.5.3
|
Evidence of the level, duration and consistency of control or protection or other intended effects of the plant protection product for each proposed use
|
|
|
Where based on extrapolation from and comparisons with authorized uses of the product concerned, a detailed explanation of the rational used. In addition, where based on extrapolation from and comparisons with authorized uses of the product concerned in other Member States, a copy of the label for the authorized product should be provided, where relevant, with a translation of the text. Where based on extrapolation from and comparisons with authorised uses of a similar product, details of the composition of the products concerned (content of each active substance) and as to the nature of the formulation (WP, EC, etc.) should be provided.
|
|
|
Where based wholly, or in part, on the results of testing in accordance with the requirements of Annex III and the Seventh Schedule, the data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
4.5.4
|
Evidence as to the possible occurrence of effects on the yield or quality of treated plants or plant products
|
|
|
Where based on extrapolation from and comparisons with authorized uses of the product concerned, a detailed explanation of the rational used. In addition, where based on extrapolation from and comparisons with authorized uses of the product concerned in other Member States, a copy of the label for the authorized product should be provided, where relevant, with a translation of the text. Where based on extrapolation from and comparisons with authorized uses of a similar product, details of the composition of the products concerned (content of each active substance) and as to the nature of the formulation (WP, EC, etc.) should be provided.
|
|
|
Where based wholly, or in part, on the results of testing in accordance with the requirements of Annex III and the Seventh Schedule, the data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
4.5.4.1
|
Evidence relating to the possible occurrence of taint or odour or other quality aspects of plants or plant products after treatment with the plant protection product, for each proposed use
|
|
|
Where based on extrapolation from and comparisons with authorized uses of the product concerned, a detailed explanation of the rational used. In addition, where based on extrapolation from and comparisons with authorized uses of the product concerned in other Member States, a copy of the label for the authorized product should be provided, where relevant, with a translation of the text. Where based on extrapolation from and comparisons with authorized uses of a similar product, details of the composition of the products concerned (content of each active substance) and as to the nature of the formulation (WP, EC, etc.) should be provided.
|
|
|
Where based wholly, or in part, on the results of testing in accordance with the requirements of Annex III and the Seventh Schedule, the data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
4.5.4.2
|
Where relevant, evidence relating to the possible occurrence of adverse effects after treatment with the plant protection product on transformation processes or on the quality of their products
|
|
|
Where based on extrapolation from and comparisons with authorized uses of the product concerned, a detailed explanation of the rational used. In addition, where based on extrapolation from and comparisons with authorized uses of the product concerned in other Member States, a copy of the label for the authorized product should be provided, where relevant, with a translation of the text. Where based on extrapolation from and comparisons with authorized uses of a similar product, details of the composition of the products concerned (content of each active substance) and as to the nature of the formulation (WP, EC, etc.) should be provided.
|
|
|
Where based wholly, or in part, on the results of testing in accordance with the requirements of Annex III and the Seventh Schedule, the data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
4.5.4.3
|
Where treated plants or plant products are likely to be stored, evidence relating to the possible occurrence of yield reduction or loss in storage
|
|
|
Where based on extrapolation from and comparisons with authorised uses of the product concerned, a detailed explanation of the rational used. In addition, where based on extrapolation from and comparisons with authorized uses of the product concerned in other Member States, a copy of the label for the authorized product should be provided, where relevant, with a translation of the text. Where based on extrapolation from and comparisons with authorized uses of a similar product, details of the composition of the products concerned (content of each active substance) and as to the nature of the formulation (WP, EC, etc.) should be provided.
|
|
|
Where based wholly, or in part, on the results of testing in accordance with the requirements of Annex III and the Seventh Schedule, the data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
4.5.4.4
|
Evidence relating to the possible occurrence of phytotoxicity after treatment with the plant protection product, for each proposed use
|
|
|
Where based on extrapolation from and comparisons with authorized uses of the product concerned, a detailed explanation of the rational used. In addition, where based on extrapolation from and comparisons with authorized uses of the product concerned in other Member States, a copy of the label for the authorized product should be provided, where relevant, with a translation of the text. Where based on extrapolation from and comparisons with authorized uses of a similar product, details of the composition of the products concerned (content of each active substance) and as to the nature of the formulation (WP, EC, etc.) should be provided.
|
|
|
Where based wholly, or in part, on the results of testing in accordance with the requirements of Annex III and the Seventh Schedule, the data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
4.5.4.5
|
Evidence relating to the possible adverse effects of a proposed treatment with the plant protection product on succeeding crops
|
|
|
Where based on extrapolation from and comparisons with authorized uses of the product concerned, a detailed explanation of the rational used. In addition, where based on extrapolation from and comparisons with authorized uses of the product concerned in other Member States, a copy of the label for the authorized product should be provided, where relevant, with a translation of the text. Where based on extrapolation from and comparisons with authorized uses of a similar product, details of the composition of the products concerned (content of each active substance) and as to the nature of the formulation (WP, EC, etc.) should be provided.
|
|
|
Where based wholly, or in part, on the results of testing in accordance with the requirements of Annex III and the Seventh Schedule, the data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
4.5.4.6
|
Evidence relating to the possible adverse effects of a proposed treatment with the plant protection product on plants or plant products to be used for propagation
|
|
|
Where based on extrapolation from and comparisons with authorized uses of the product concerned, a detailed explanation of the rational used. In addition, where based on extrapolation from and comparisons with authorized uses of the product concerned in other Member States, a copy of the label for the authorized product should be provided, where relevant, with a translation of the text. Where based on extrapolation from and comparisons with authorized uses of a similar product, details of the composition of the products concerned (content of each active substance) and as to the nature of the formulation (WP, EC, etc.) should be provided.
|
|
|
Where based wholly, or in part, on the results of testing in accordance with the requirements of Annex III and the Seventh Schedule, the data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
5
|
DATA AND INFORMATION RELATING TO RESIDUES IN TREATED PRODUCTS, FOOD OR FEED FOLLOWING USE AS PROPOSED — WHERE REQUIRED
|
|
5.1
|
Evidence of compliance with existing MRLs
|
|
5.1.1
|
On the basis of extrapolation from data relevant to other crops or plant products.
|
|
5.1.2
|
On the basis of supervised trial data.
|
|
|
The data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
5.2
|
Data and information to support the establishment of a Provisional MRL
|
|
5.2.1
|
On the basis of extrapolation from data relevant to other crops or plant products.
|
|
5.2.2
|
On the basis of supervised trial data.
|
|
|
The data must be reported as specified in Part 2 of the Fourth Schedule.
|
|
| |
PART 3
|
| |
Regulation 16(2)
|
| |
GENERAL RULES RELATING TO COMPARABILITY, EXTRAPOLATION, GROUP TOLERANCES AND DATA REQUIREMENTS32
|
| |
CONTENTS
|
| | |
1
|
Introduction
|
|
2
|
General principles
|
|
2.1
|
Least favourable trial conditions
|
|
2.2
|
Definition of comparability
|
|
2.3
|
Comparative trials
|
|
2.4
|
Consideration of existing information and experience
|
|
2.5
|
Properties of active ingredients
|
|
2.6
|
Non-relevant residues
|
|
3
|
Changes in the trial parameters
|
|
3.1
|
Formulation
|
|
3.2
|
Application rate
|
|
3.3
|
Number of applications
|
|
3.4
|
Application method
|
|
3.5
|
Timing of application; Pre-harvest Interval
|
|
3.6
|
Area of application
|
|
3.7
|
Simultaneous changes in more than one trial parameter
|
|
4
|
Comparable climatic zones/weather influences
|
|
5
|
Comparable residue behaviour in different crops
|
|
5.1
|
Basic requirements (number and type of trials)
|
|
5.2
|
Test series/values at harvest
|
|
5.3
|
Inference of group tolerances
|
|
5.4
|
Residue situations
|
|
5.4.1
|
High residue risk
|
|
5.4.2
|
Medium residue risk
|
|
5.4.3
|
Low residue risk
|
|
32 The text comprises Appendix D of Commission Document 2946/VI/93: Study, prepared By Dr Jörg-Rainer Lundehn for the Commission of the European Communities, Guidelines for the Establishment of Community Maximum Residue Levels (MRLs) of Plant Protection Products in Food and Feedstuffs of Plant and Animal Origin, January 1993.
|
|
5.4.4
|
Special case — rotational crops
|
|
Annex II
|
"Major Crops" list
|
|
Figure 2
|
Division of France into two regions
|
|
Figure 3
|
Comparison of 'normal' and 'reverse' test series Introduction
|
|
1.
|
Introduction
|
|
|
On the basis of existing knowledge and findings it can be assumed that, taking the least favourable trial conditions, the residue behaviour in/on plants or plant products is, under certain circumstances, comparable. In such cases, existing knowledge about the residue behaviour in one situation can be transferred to another situation, and the scale of the trials for the comparable situation can be reduced, or trials may even be completely unnecessary.
|
|
|
In the following text guidelines, residue situations which are assumed to be comparable on the basis of existing information currently available are described, and recommendations are made as to the type and scale of the residue trial results which have to be submitted. New findings may result in a change of assessment of comparability.
|
|
|
A number of rules are based on conventions and considerations of plausibility.
|
|
|
Naturally, it is not possible to describe all conceivable situations, and even in established cases special factors frequently intervene which are difficult to evaluate.
|
|
|
The responsibility of the applicant to submit all the data necessary for the evaluation remains unaffected.
|
|
2
|
General principles
|
|
2.1
|
Least favourable trial conditions
|
|
|
When testing residue behaviour, the principle is to choose the trial conditions which under realistic circumstances would be the least favourable. The 'least favourable trial conditions' are those which under the given circumstances produce what would probably be the worst residue situation (e.g. maximum number of possible applications, highest prescribed dosage), while also being representative (spreading of the trials over more than one — usually two — growing seasons, consideration of main growing regions, influence of varieties, standard application methods and times).
|
|
|
It is mainly the results of controlled residue trials which form the basis for the estimation of maximum residue levels of plant protection products on products of plant and animal origin. Maximum residue levels are set as high as necessary on the basis of application as provided for authorization, taking into account imports and the demands of international trade, and as low as possible for reasons of preventive health care, and never under any circumstances higher than can be justified on toxicological grounds. In individual cases, the result of this may be that if the least favourable application conditions provided for in the authorization cease to apply, then the maximum residue limit may be set on the basis of the next most unfavourable conditions. In this case, results of residue trials must always be submitted if it can be supposed with good reason that on consideration of these next most unfavourable residue conditions the maximum residue limit might possibly be reduced by at least one category.
|
|
2.2
|
Definition of comparability
|
|
|
Residue levels for relevant different harvested crops are considered to be comparable if:
|
|
1.
|
( a ) assuming a standard distribution of data the respective 'mean to one-sigma-limit' ranges overlap; or
|
|
|
( b ) assuming a non-standard distribution of data the respective 'median to upper percentile (75 % tile)' ranges overlap;
|
|
|
and
|
|
2.
|
if the resulting recommended maximum residue levels according to the recommended calculation procedure fall, into the same or a neighbouring maximum residue limit category after rounding up or down to the nearest maximum residue limit category. For this purpose the following methods of calculation are used:
|
|
|
(a) assuming a standard distribution of data:
|
|
|
 = mean residue = mean residue
|
|
 max =R + kxs max =R + kxs
|
k= factor
|
|
|
s = standard deviation
|
|
|
(b) assuming a non-standard distribution of data:
|
|
Rcalculated = 2 x R0.75
|
R0.75 = 75 % quintile
|
|
| | |
|
For the source of the definitions, see also Mitteilungen aus der Biologischen Bundesanstalt für Land- und Forstwirtschaft (Bulletin of the Federal Biological Research Centre for Agriculture and Forestry), No. 263, July 1990.
|
|
2.3
|
Comparative trials
|
|
|
Comparative trials at a single trial site must be organised in such a way that to the greatest possible extent genuinely comparable conditions can be expected. Owing to largely unpredictable weather conditions, trials at several different sites, with a sufficient regional spread, are necessary as a general principle. The number of trial sites depends on the question under investigation, but as a rule it should not be less than four. The trials are to be carried out under normal practical conditions of agriculture. Under special circumstances, however, it may also be appropriate to carry out trials under controlled conditions, e.g. in climate-controlled chambers or greenhouses, in which the factors which influence residue behaviour can be controlled.
|
|
2.4
|
Consideration of existing information and experience;
|
|
|
Already existing information and experience, and the systematic evaluation of these results often make it possible to reduce the number of trials needed, or to answer the question under investigation without carrying out further trials. When evaluating trial results, the existing information should always be considered and evaluated. Experience shows that where residues occur, approximately 25 trials from outdoor are generally sufficient in order to be able to judge with sufficient certainty what the residue behaviour of an active ingredient would be in a particular comparable situation.
|
|
2.5
|
Properties of active ingredient (stability, volatility, mode of action, uptake and distribution)
|
|
|
It can be assumed, and this has also been confirmed in a number of cases on the basis of existing information, that the residue behaviour of different active ingredients is comparable. This presupposes that sufficient information (i.e. metabolism, physical chemical properties, residue results) already exists for these active ingredients. If comparability is assumed, then this must be carefully substantiated with the existing information.
|
|
2.6
|
Non-relevant residues
|
|
|
Residues are considered to be non-relevant in the sense of the following rules if their content in the harvested product is below the limit of determination (i.e. generally between 0.01 and 0.05 mg/kg). This is often the case with the early application (e.g. applications in autumn or spring) of herbicides, applications of non-systemic insecticides and fungicides on fruits prior to flowering, and seed dressings.
|
|
|
The fact that no detectable residues occur or residues are non-relevant is often due to the properties of the active ingredient, the type and timing of application, the rate of application, and the results of metabolic studies and studies of the plant's uptake and distribution of the compound.
|
|
|
If no detectable residues occur under the least favourable trial conditions, no further trial results are required if trial conditions are changed to less unfavourable ones.
|
|
|
In situations where non-relevant residues can be expected with a high degree of probability, then as an exception to the basic rule (spreading of the trial over two years), all the trials may be carried out within one growing season. If, however, contrary to expectations relevant detectable residues should be found after all, then further results must be obtained in a second growing seasons.
|
|
3
|
Changes in the trial parameters
|
|
|
The following rulings presuppose that in each case the original situation is sufficiently well documented.
|
|
|
If, when changes are made to the trial parameters, the obtaining of further residue results is considered not to be necessary, then thorough justification for this must be submitted. A justification could be, for instance, that existing trial results show that relevant residues are unlikely to occur.
|
|
3.1
|
Changes in formulation
|
|
|
As a basic principle, residue trials must be carried out using the formulation to which the authorization applies, or for which the application has been made. If there is a significant change in formulation, therefore, new residue trials are, in principle, necessary. It has proved sufficient to carry out four comparative trials on each crop selected. Data are not needed for all crops, but should be generated for approximately 3 major crops representative of the crop groups which may be treated, e.g. a leafy crop, a root crop, a soft fruit, a tree fruit, a seed crop etc. The trials should preferably be carried out on crops which would be expected to show high levels of residues. The timing of treatment is also important in this situation. Where treatments are made to the soil the formulation is not important and where treatment is to a very young crop the effect of co-formulants is likely to be minimal. In cases of minor changes in formulation, which would not be expected to have any influence on efficacy and residue behaviour, additional trials may be waived.
|
|
|
Experience shows that EC, WP and SC formulations produce comparable residues, especially if last application is more than seven days prior to harvest,
|
|
3.2
|
Changes in application rate
|
|
|
In order to encompass the least favourable trial conditions, the trials must as a matter of principle be carried out using the highest rate (e.g. kg/ha) of application. In the case of active ingredients which act via the soil (e.g. pre-emergence herbicides), the application rate appropriate for the particular type of soil should be used. In the case of increases or reductions of up to 25% in the rate of application of the active ingredient under otherwise identical conditions, experience suggests that the residue results can be assumed to be comparable.
|
|
3.3
|
Changes in number of applications
|
|
|
In order to encompass the least favourable trial conditions, the trials must as a matter of principle be carried out using the maximum number of application provided for in the authorization. It is generally the last application prior to harvest which are crucial to residue behaviour in the harvested crop. The number of applications prior to flowering, on the other hand, is generally of lesser importance. Even in the case of relatively stable active ingredients, the residue results can be assumed to be comparable if the number of applications after flowering are increased or reduced by not more than 25% (e.g., 4 1 or 8 2 applications).
|
|
3.4
|
Changes in application method
|
|
|
Different application methods, such as spraying, drenching, dusting, misting and granule spreading, will as a rule not produce comparable residue results, and must therefore be documented separately. The results from normal spraying and low-volume spraying are often comparable for a comparable rate of application for the active ingredient per ha. However where both, low volume and normal spray applications, are the usual methods, both methods of application ought to be documented according to standard application practice in the basic data set submitted.
|
|
3.5
|
Changes in timing of application; changes in pre-harvest interval
|
|
|
The stage of development of the crop and the time intervals between applications are important factors influencing the level of residues. Because for the setting of maximum residue limits the least favourable residue situation is the determining factor, applications at later stages of development will encompass applications made at earlier stages of development, just as applications at shorter intervals before harvesting will encompass applications at longer intervals before harvesting (but note Section 2.1, paragraph 3).
|
|
|
In the case of changes in pre-harvest interval of not more than 25%, experience has shown that the residue results can be assumed to be comparable.
|
|
3.6
|
Area of application (outdoors, under glass, in store, under foil)
|
|
|
The results of outdoor trials are not normally comparable with the results of trials carried out in other areas of application. The climatic conditions, above all, under glass, under foil, or in climate controlled chambers or in stores, but also the other parameters which differ from those in outdoor trials, generally create a markedly worse residue situation than that found in outdoor testing. Therefore, separate full-scale studies are necessary for each area of application.
|
|
3.7
|
Simultaneous changes in several trial parameters
|
|
|
The 25 % rule mentioned in Sections 3.2, 3.3 and 3.5 for purposes of comparability only applies where just one of the parameters is changed. Where more than one parameter is changed at the same time, the effects may be cumulative, or may cancel each other out.
|
|
|
Thus, for example, increasing the application rate by 20 % while at the same time reducing the number of applications from 4 to 3 will probably result in a comparable residue behaviour. If, however, the number of applications were instead increased from 4 to 5, it would be quite obvious that the residue behaviour would no longer be comparable. The stability of the active ingredient and the timing of applications and intervals between applications naturally also play a crucial part in this.
|
|
|
If more than two trial parameters are changed at the same time, experience suggests that it is then no longer possible to assume a comparable residue behaviour with any sufficient degree of certainty.
|
|
4
|
Comparable climatic zones/weather influences
|
|
|
It is assumed that for the carrying out of residue trials the climatic conditions and weather influences in each of the two regions described below are comparable:
|
|
|
( a ) Northern and Central Europe
southern Sweden, southern Norway, Denmark, Great Britain, Ireland, northern and central France, Belgium, The Netherlands, Luxembourg, Germany, Poland, Czechoslovakia, Austria, Switzerland.
|
|
|
( b ) Southern Europe and the Mediterranean
Spain, Portugal, southern France, Italy, Greece, Yugoslavia, Turkey, Bulgaria, Rumania.
|
|
|
With regard to the division of France between the two regions, see Figure 2.
|
|
|
Data from different countries within the same region may reflect different cultural practice and they might be rejected as irrelevant on that basis. Results from regions which are not climatically comparable cannot in general serve as a total substitute for trials carried out in comparable regions. They do, however, add to knowledge about the residue behaviour of the active ingredient.
|
|
|
Extensive data from several climatic zones (e.g. in the USA) may, however, in individual cases provide a perfectly adequate basis on which to evaluate the residue situation in the member states of the EC.
|
|
5
|
Comparable residue behaviour in different crops
|
|
5.1
|
Basic requirements (number and type of trials)
|
|
|
The requirement for each of the two regions (8 trials for major crop, 4 trials for minor crop) represents a basic requirement which in principle must be adhered to, but which may be amended in individual cases depending on the given situation and other prior knowledge (properties of the active ingredient, e.g. metabolic studies, stability, rates of application, method of application, type and scale of existing residue trial results from other crops or other countries). As a result of this it may be that sufficient conclusions can be drawn for the evaluation of residue behaviour from a smaller number of trial results. In some cases, on the other hand, it may be necessary to carry out a larger number of trials in order to eliminate any points of confusion. In all cases, all the available facts must be considered by an experienced expert in order to make the evaluation.
|
|
|
For the list of 'major crops' see Annex 2 of this Schedule.
|
|
5.2
|
Test series/values at harvest
|
|
|
Test series are residue trials with samples taken usually on five occasions, of which two are often fixed times: the day of the final application and the time of harvesting. In all cases the proposed pre-harvest interval must be taken into account when taking samples.
|
|
|
Despite higher trial and analysis costs, test series have an advantage over values at harvest (the taking of samples at the time of harvesting) in that they provide an opportunity of assessing the residue behaviour over a period of time, and from the dissipation curve obtained in this way it is possible to make a relatively reliable estimate of residues at the time of harvesting (e.g. by identifying outliers and/or the important influencing factors, such as relative decrease in residues as a result of plant growth and the effects of the weather (temperature, precipitation)). Besides through test series it is also possible to monitor initial deposits, and hence also the proper application of the product.
|
|
| |
|
| |
Figure 2
|
| |
DIVISION OF FRANCE INTO THE TWO REGIONS DESCRIBED IN SECTION 4
|
| |
|
| | |
|
From the above it will be clear that test series are particularly appropriate and necessary in cases where a pre-harvest interval has to be determined, or where the possibility cannot be ruled out that various different pre-harvest intervals may be considered.
|
|
|
If a plant protection product is used several times during the growing season of a crop, a good method, and one which is recommended, has proved to be to take the first sample immediately prior to the final application; this makes it possible to ascertain the influence of the previous applications on the level of residues.
|
|
|
Under certain circumstances (e.g. in the case of applications of a plant protection product in cereals prior to flowering), owing to the fact that the sample material is not comparable (green matter, ears, grain/straw) it is sufficient to carry out shortened test series consisting of less than five (normally three) sampling times.
|
|
|
Experience has also shown that in some circumstances the knowledge gained from about 2-3 value-at-harvest results from different trials may be comparable with that gained from a single test series.
|
|
|
As already stated, in a normal test series samples are taken, following treatment, from a single treated plot at appropriate intervals right up to harvesting. Alternatively, it is possible to carry out so called 'reverse test series', and this is especially recommended where the pre-harvest interval may range over a relatively long period of time. In a reverse test series, the product is applied to neighbouring plots at intervals corresponding to the possible treatment period prior to harvesting, and samples are taken from all the plots at the same time, at harvesting. For an explanation of 'reverse test series', see Figure 3.
|
|
5.3
|
Inference of group tolerances
|
|
|
Inference of group tolerances is carried out in three steps:
|
|
1.
|
Collection of residue data for the relevant representative crops of the group.
|
|
2.
|
Testing of the results for comparability according to the procedure described under 2.2
|
|
3.
|
Decision
|
|
|
1st case: comparability given calculation of group tolerance on the basis of all the available data
|
|
|
2nd case: no comparability.
|
|
|
— setting of different maximum residue limits for the individual crops; no setting of a group tolerance at this stage
|
|
|
— studies of further crops if necessary (i.e. if GAP exists for that crops)
|
|
|
Group tolerances will normally only be discussed if Good Agricultural Practice (GAP) for the group-crops is comparable.
|
|
5.4
|
Residue situations (definition and data requirements)
|
|
|
The comparability of residue behaviour in different crops is dependent on a series of factors which influence the level of residues and their increase and decrease over a period of time. Therefore, under a different set of conditions (residue risk and data requirements), comparability will also differ.
|
|
|
Below, three different cases will be distinguished:
|
|
|
— residue situations which are difficult to anticipate (= high residue risk)
|
|
|
— residue situations which can be anticipated to some extent (= medium residue risk)
|
|
|
— situations in which usually few or no residues occur ( = low residue risk)
|
|
| |
Figure 3
|
| |
Comparison of 'normal' and 'reverse' test series
|
| | |
|
Normal test series
|
|
one trial plot
|
|
samples taken on five occasions
|
|
i.e. 1 trial plot:
|
|
treatment and sampling carried out on the single plot at intervals of time, e.g. 0, 7, 14, 21 and 28 days after last treatment.
|
|
Reverse test series
|
|
|
|
1st trial plot
|
|
e.g.
treatment 28 days before harvest
|
|
|
|
2nd trial plot
|
|
treatment 21 days before harvest
|
|
|
|
3rd trial plot
|
|
treatment 14 days before harvest
|
|
|
|
4th trial plot
|
|
treatment 7 days before harvest
|
|
|
|
5th trial plot
|
|
treatment immediately before harvest
|
|
|
i.e.— 5 neighbouring trial plots
|
|
|
— treatment at intervals of time (28, 21, 14, 7, 0 days before harvest on the appropriate plot)
|
|
|
— Sampling on all plots on the same day at the time of harvesting.
|
|
5.4.1
|
High residue risk (= residue situations which are difficult to anticipate)
|
|
|
Definition:
|
|
|
It is not possible, or possible only to a limited extent (e.g. if there has already been extensive experience with the active ingredient) to anticipate the residue behaviour in the harvested crop as regards residue level and changes in residues over the course of time. In particular, cases where the harvested crop is not directly contaminated by treatment, but where uptake by the plant and translocation to edible parts of the plant are effectively or very effectively (example: soil applications of aldicarb).
|
|
|
Data requirements: 8 test series for major crops 4 test series for minor crops (basic requirements per region)
|
|
|
Generally speaking:
|
|
|
In cases where a high residue risk must be assumed, a relatively large number of result is required in order to evaluate residue behaviour, and comparability/ transferability and group tolerances can only be inferred in relatively few cases.
|
|
|
The fact that in these cases residue behaviour can be anticipated only to a very limited extent makes it extremely difficult to predict comparability/transferability from one crop to another and to define the preconditions for setting group tolerances.
|
|
|
Some reference points where comparability might be given even in high-residue risk situations are provided by the cases described below, e.g.:
|
|
grapefruits
apples
peaches
table grapes
onions
sugar beet
turnips
carrots
cauliflower
red cabbage
dry peas
rape seed
wheat
|
oranges
pears
apricots
wine grapes
shallots
fodder beet
swedes
parsley root
broccoli
white cabbage
dry beans
linseed
triticale
|
rye
|
|
|
Explanations:
|
|
a
|
b
|
This means that on the basis of practical experience, the transferability of residue results both from a to b and from b to a are admissible.
|
|
Group, Group I, Group II, Group III, etc.
|
Are the crop groups set out in the Annex to Council Directive 90/642/EEC, as amended by Council Directive 93/58/EEC.
|
|
| | |
|
The root systems of the different plants and knowledge about the uptake and distribution of the active ingredient by the plants also provide important reference points. Finally, the more data already exist for different crops, the more easily will experienced experts be able to infer comparability/transferability and group tolerances. The spread of residues (mean value, median, standard deviation, 75% quantile) and the level of residues also play an important role.
|
|
5.4.2
|
Medium residue risk (= residue situations which can be anticipated to some extent)
|
|
|
Definition:
|
|
|
The harvested crop, or parts of the harvested crop, are contaminated during treatment (e.g. applications to the harvested crop, or application after flowering of fruits to shortly before harvest, or applications shortly after harvest and in store (storage protection)). The level of residues and changes in them over the course of time can be anticipated to a limited extent on the basis of the morphology of the harvested crop and predictable factors (decrease as a result of growth (dilution), evaporation, metabolisation), taking into account the existing findings.
|
|
|
Generally speaking:
|
|
|
In cases where a medium residue risk can be assumed, the basic set of data is sufficient, for the evaluation of residue behaviour in individual crops. Where mutual transferability can be assumed, the trials can also be spread among the comparable crops (e.g. 4 trials on apples and 4 trials on pears).
|
|
|
Data requirements:
|
|
|
8 trials (of which 4 test series) for major crops
|
|
|
4 trials (of which 2 test series) for minor crops (basic requirements per region)
|
|
|
Post-harvest treatments (including storage protection):
|
|
|
In the case of post-harvest treatments and applications for storage protection, variability in the residues is smaller owing to the lack of climatic influences. It is therefore generally sufficient to carry out 4 trials in one year. In this case it is not necessary to spread the trials over the two European regions described previously. 2 of the trials should however be in the form of test series. Sampling must take account of the variability of residues in the store. Furthermore, separate studies must be carried out for different store types and for the different types of treatment (dipping, spraying, misting, fumigation). In the case of treatments in closed rooms using stable active ingredients it is sometimes also possible to calculate the maximum residues from the quantity of the active ingredient concerned and the quantity of the product treated.
|
|
| |
Explanations:
|
| | |
|
=
|
transferability is asmissible from a to b
|
|
|
=
|
transferability is admissible both from a to b and from b to a
|
|
|
=
|
inference of a group tolerance
|
|
| | |
Group, Group I, Group II, Group III, etc.
|
Are the crop groups set out in the Annex to Council Directive 90/642/EEC, as amended by Council Directive 93/58/EEC.
|
|
| |
1. Fruit
|
| |
I Citrus fruits
|
| |
|
| | |
|
Explanatory notes:
|
|
|
Residue studies on grapefruits, oranges and pomelos are mutually transferable. Residue studies on lemons, mandarins and limes are also mutually transferable. For the setting of individual tolerances, 8 trial results are required for the major crops oranges, lemons and mandarins, and the results for comparable crops are taken into account. For grapefruits, pomelos and limes, as minor crops for the EC-member states, 4 trial results would suffice for applications in the European Community. For the setting of import tolerances, a total of 8 trial results are required for grapefruits and pomelos (see also the List of Major Crops in the Annex) because on a worldwide basis grapefruit and pomelos are assumed to be major crops.
|
|
|
For the setting of a maximum residue limit for the sub-group grapefruits, oranges and pomelos, a total of 8 trial results from Southern Europe are sufficient. The same applies to the sub-group lemons, mandarins and limes. The trials may be carried out on any one of the crops in the appropriate sub-group. It is also possible, however, to spread the trials over more than one or all of the crops in the sub-group. It is recommended that the studies should be concentrated on the major crops. To set a group tolerance for the whole of Group I, 8 trial results are required for each of the two sub-groups. For post-harvest treatments (including storage protection) 4 trials are required for each of the two sub-groups. These explanations also apply, unless stated otherwise, to the corresponding cases below.
|
|
|
II Tree nuts
|
|
| |
|
| |
|
| | |
|
Explanatory notes:
|
|
|
For the setting of a group tolerance, 16 trial results are required. For post-harvest treatments only 8 trial results are required.
|
|
|
III Pome fruit
|
|
| |
|
| |

|
| | |
|
Explanatory notes:
|
|
|
For the setting of a group tolerance, 8 trial results are required. For post-harvest treatments only 4 trial results are required.
|
|
| |
|
| |
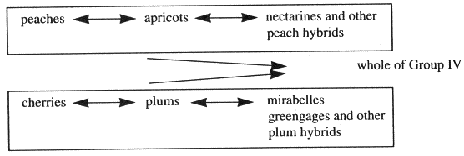
|
| | |
|
Explanatory notes:
|
|
|
To set a group tolerance for the whole of Group IV, 8 trial results are required for each of the two sub-groups. For post-harvest treatments only 4 trial results are required for each of the two sub-groups.
|
|
|
VBerries and small fruit
|
|
|
( a ) Table and wine grapes
|
|
| |
|
| | |
|
( b ) Strawberries
|
|
|
No transferability, therefore 8 trial results or 4 trial results for post-harvest treatments.
|
|
|
( c ) Cane fruit (other than wild)
|
|
| |

|
| | |
|
( d ) Other small fruits and berries (other than wild)
|
|
| |
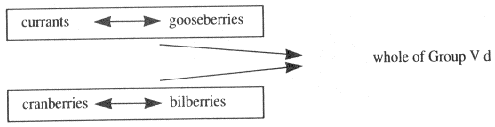
|
| | |
|
( e ) Wild berries and wild fruits (eg., in the case of application in forests)
|
|
| |
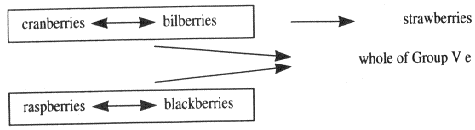
|
| | |
|
Explanations:
|
|
|
Studies carried out on cultivated forms are under certain circumstances transferable to wild forms (with the exception of strawberries). Because of the small proportion of total consumption, half the basic data is sufficient for wild berries, which are all classed as minor crops — i.e. a total of 4 trial results from each sub-group are sufficient for setting a group tolerance (except for wild strawberries).
|
|
|
If for each of 4 different sub-groups out of the Berries and Small Fruit Group at least 6 study results exist showing a comparable residue behaviour, then the setting of a group tolerance for the whole of Group V can be discussed.
|
|
|
VIMiscellaneous fruits
|
|
|
At present there is no known transferability, therefore 4 or 8 trial results per crop required, depending on whether it is a minor or a major crop.
|
|
2.
|
Vegetables
|
|
I
|
Root and tuber vegetables (applications prior to harvest)
Not relevant here, see rulings on high or low residue risk.
|
|
|
Root and tuber vegetables (post-harvest treatments and storage protection)
|
|
| |
|
| | |
|
Explanatory notes:
|
|
|
Experience suggests that studies on 2 crops (4 trials for each crop) including carrots from different sub-groups are sufficient for setting a group tolerance for the whole of Group I.
|
|
|
II Bulb vegetables
|
|
| |
|
| | |
|
Explanatory notes:
|
|
|
For the setting of a group tolerance for Group II, 12 trial results are required (8 trial results for the sub-group onions, shallots and garlic, and 4 trial results for spring onions).
|
|
| | |
|
IIIFruiting vegetables
|
|
|
( a ) Solanaceae
|
|
| |
|
| |
|
| | |
|
( b ) Cucurbits — edible peel
|
|
| |
|
| |

|
| | |
|
( c ) Cucurbits inedible peel
|
|
| |
|
| |

|
| |
|
| |
|
| |
|
| | |
|
i.e. 4 trial results for grain maize (milky stage) are sufficient for setting a maximum residue limit for sweet corn.
|
|
|
Explanatory notes:
|
|
|
If sufficient trial results exist for 2 different sub-groups out of Groups a), b) and c) and for sweet corn/maize showing a comparable residue behaviour, then the setting of a group tolerance for the whole of Group III can be discussed.
|
|
|
IVBrassica vegetables
|
|
|
( a ) Flowering brassicas
|
|
| |
|
| |

|
| |
|
| |
|
| |
|
| |
|
| |
|
| |

|
| |
|
| | |
|
No transferability, therefore 4 trial results
|
|
|
Explanatory notes:
|
|
|
A minimum of 6 studies for each of sub-groups (a), (b) and (c) and 4 trial results for kohlrabi are sufficient for setting a group tolerance for brassica vegetables (Group IV), if a comparable residue behaviour is found between the sub-groups. For post-harvest treatments only 4 studies for each of the sub-groups are required.
|
|
|
V Leafy vegetables and fresh herbs
|
|
|
( a ) Lettuces and similar
|
|
| |
|
| |
|
| | |
|
( b ) Spinach and similar
|
|
| |
|
| |

|
| | |
|
( c ) Watercress
No transferability, therefore 4 trial results required
|
|
|
( d ) Witloof
No transferability, therefore 8 trial results required
|
|
|
( e ) Fresh herbs
|
|
|
At present there is no known transferability, therefore currently 4 trial results for each crop required.
|
|
|
Explanatory notes:
|
|
|
If sufficient trial results are available showing a comparable residue behaviour for lettuce, lamb's lettuce and spinach or the comparable crops in each case, then the setting of a group tolerance for Group V can be discussed.
|
|
|
VI Legume vegetables (fresh)
|
|
| |
|
| |

|
| |
|
| |
|
| | |
|
Explanatory notes:
|
|
|
For the setting of a group tolerance, studies on celery and leeks are required.
|
|
|
VIII Fungi
|
|
|
( a ) Cultivated fungi
|
|
| |
|
| | |
|
Explanatory notes:
|
|
|
For the setting of a maximum residue limit for cultivated fungi, 8 trial results on mushrooms and 4 trials on another species of fungus (e.g. oyster mushrooms) are required.
|
|
|
( b ) Wild mushrooms
|
|
| |

|
| | |
|
Explanatory notes:
|
|
|
For the setting of a maximum residue limit for wild mushrooms, 4 trial results for each of the two sub-groups are sufficient.
|
|
|
3. Pulses
|
|
|
( a ) Applications prior to harvesting
|
|
| |

|
| | |
|
( b ) Applications after harvesting or for storage protection
|
|
| |

|
| | |
|
Explanatory notes:
|
|
|
For the setting of a maximum residue limit for pulses (application after harvesting of for storage protection) 4 trials carried out in one year are generally sufficient. For application prior to harvesting 8 trials on beans/peas and 4 results on lentils have to be worked out.
|
|
|
4. Oil seeds
|
|
| |
|
| | |
|
Explanatory notes:
|
|
|
If a minimum of 4 trial results showing a comparable residue behaviour are available for each of 3 different sub-groups (including rape seed) out of Group 4, then the setting of a group tolerance for the whole of Group 4 can be discussed.
|
|
|
5. Potatoes
|
|
|
(a) application prior to harvest
|
|
|
Not relevant here, see rulings on high or low residue risk.
|
|
|
(b) post-harvest treatments
|
|
|
Explanatory notes:
|
|
|
It is generally sufficient to carry out 4 trials in one year.
|
|
|
6. Tea
|
|
|
No transferability, therefore 8 trial results for the setting of an import tolerance.
|
|
|
7. Hops
|
|
|
No transferability, therefore 8 trial results.
|
|
|
8. Cereals
|
|
|
( a ) Applications prior to harvest
|
|
| |
|
| | |
|
Explanatory notes:
|
|
|
A minimum of 6 study results for each of wheat, barley, maize and rice are sufficient for the setting of a maximum residue limit for cereals if there is a comparable residue behaviour.
|
|
|
( b ) Applications after harvesting and for storage protection
|
|
| |
|
| | |
|
Explanatory notes:
|
|
|
A minimum of 4 study results for each of wheat, maize and rice are sufficient for the setting of a maximum residue limit for cereals if there is a comparable residue behaviour.
|
|
5.4.3
|
Low residue risk (Situations in which usually low or no residues occur)
|
|
|
Definition:
|
|
|
Situations in which on the basis of experience no residues, or else only very low residues (
|
|
|
In this group of cases, several different situations with regard to data requirements, comparability and determining of group tolerances are distinguished.
|
|
|
IPlants not present at the time of treatment
|
|
|
( a ) Application to the soil
|
|
(e.g. applications
|
—
|
pre-sowing
|
|
|
—
|
pre-planting
|
|
|
—
|
pre-emergence)
|
|
|
( b ) Seed-dressing treatments
|
|
|
IIPlants present at the time of treatment
|
|
|
( a ) Harvested crop not formed or barely formed:
|
|
(e.g. early applications
|
—
|
post-emergence
|
|
|
—
|
post-planting
|
|
|
—
|
pre-flowering)
|
|
|
( b ) Harvested crop formed:
|
|
|
1. Harvested crop grows totally or largely underground
|
|
|
2. Harvested crop grows above the ground, but owing to the method of application comes only into little or no contact with the product
|
|
(e.g
|
—
|
applications of herbicides in crops growing high off the ground (fruit, vines, hops)
|
|
|
—
|
treatments between the rows
|
|
|
—
|
under leaf treatments
|
|
|
Generally speaking:
|
|
|
In cases where a low residue risk can be assumed, experience shows that fewer residue data are required than in cases of medium and high risk. Transferability and group tolerance are more easily inferred, since often no residues (i.e. below the limit of determination) or only very low residues (i.e.
|
|
|
Within the cases and crop groups described below, residue behaviour is assumed to a large extent to be comparable, so descriptions of transferability in individual cases are not given. However, for the setting of group tolerances, certain minimum sets of data are required, and these are described below.
|
|
Data requirements:
|
6 trials (of which 2 test series) for major crops
|
|
|
4 trials (of which 1 test series) for minor crops (Basic requirement per region)
|
|
Case I
|
|
Plants not present at the time of treatment.
|
|
Case Ia
|
Applications to the soil
|
|
|
(e.g. applications
|
—
|
pre-sowing
|
|
|
|
—
|
pre-planting
|
|
|
|
—
|
pre-emergence)
|
|
|
|
|
|
|
Crop/group of crops
|
Number of trials
|
Comments
|
|
Citrus fruits
|
|
not relevant i.e. residue studies
|
|
|
|
not required because residues are not expected
|
|
Tree nuts
|
|
not relevant
|
|
Pome fruit
|
|
not relevant
|
|
Stone fruit
|
|
not relevant
|
|
Table and wine grapes
|
|
not relevant
|
|
Strawberries
|
4
|
|
|
Cane fruits
|
|
not relevant
|
|
Other small fruit and berries
|
|
not relevant
|
|
Wild fruits
|
|
not relevant
|
|
Miscellaneous fruits
|
|
not relevant
|
|
Root and tuber vegetables
|
8
|
i.e. studies on a minimum of 2 crops (one major crop and one minor crop) are necessary for the setting of a group tolerance. The studies may also be spread over several crops (= Expl. 1)
|
|
Bulb vegetables
|
6
|
i.e. studies on one major crop are sufficient for the setting of a group tolerance. The studies may also be spread over several crops (= Expl. 2)
|
|
Fruiting vegetables
|
12
|
i.e. studies on a minimum of 2 crops (one major crop and one minor crop) are necessary for the setting of a group tolerance. The studies may also be spread over several crops. The studies must be carried out on crops from different sub-groups (= Expl. 3)
|
|
Brassica vegetables
|
12
|
see Expl. 3
|
|
Leafy vegetables and fresh herbs
|
12
|
see Expl. 3
|
|
Legume vegetables (fresh)
|
6
|
see Expl. 2
|
|
Stem vegetables
|
8
|
see Expl. 1
|
|
Fungi
|
4
|
see Expl. 2
|
|
Pulses
|
6
|
see Expl. 2
|
|
|
|
If studies on legume vegetables (fresh) are available, the results are transferable
|
|
Oil seeds
|
8
|
see Expl. 1
|
|
Potatoes
|
4
|
|
|
Hops
|
4
|
|
|
Cereals:
|
|
|
|
( a ) Barley
Oats
Rye
Wheat
Triticale
Buckwheat
|
|
6
|
i.e. studies on a minimum of one crop from each of Group a) and b) are required for the setting of a group tolerance for cereals
|
|
( b ) Maize
Millet
Sorghum
Rice
|
|
6
|
|
|
Case Ib
|
Seed-dressing treatments
|
|
Crop/group of crops
|
Number of trials
|
Comments
|
|
Citrus fruits
|
|
not relevant
|
|
Tree nuts
|
|
not relevant
|
|
Pome fruit
|
|
not relevant
|
|
Stone fruit
|
|
not relevant
|
|
Berries and small fruit
|
|
not relevant
|
|
Miscellaneous fruits
|
|
not relevant
|
|
Roots and tuber vegetables
|
6
|
see Expl. 2
|
|
Bulb vegetables, including leeks
|
6
|
see Expl. 2
|
|
Fruiting vegetables
|
|
|
|
(a) Solanaceae
|
6
|
see Expl. 2
|
|
(b) Cucurbitaceae
|
6
|
see Expl. 2
|
|
(c) Sweet corn
|
|
see cereals
|
|
Brassica vegetables
|
6
|
see Expl. 2
|
|
Leafy vegetables and fresh herbs
|
12
|
see Expl. 3
|
|
Legume vegetables (fresh) and pulses
|
6
|
see Expl. 2
|
|
Stem vegetables (where relevant)
|
6
|
see Expl. 2 for leeks see bulb vegetables
|
|
Fungi, including wild forms
|
|
not relevant
|
|
Oil seeds
|
8
|
see Expl. 1
|
|
Potatoes
|
4
|
|
|
Tea
|
|
not relevant
|
|
Hops
|
|
not relevant
|
|
Cereals:
|
|
|
|
( a ) Barley
Oats
Rye
Wheat
Triticale
Buckwheat
|
|
6
|
i.e. studies on a minimum of one crop from each of Groups (a) and (b) are required for the setting of a group tolerance for cereals
|
|
( b ) Maize including sweet corn
Millet
Sorghum
Rice
|
|
6
|
|
|
Explanatory notes:
|
|
If in studies on potatoes, cereals, legume vegetables/pulses and oil seeds no detectable residues occur, then no further studies are necessary for the other crops or groups of crops. For the purposes of the studies it is generally sufficient to carry out shortened test series.
|
|
Case II
|
|
Plants present at the time of treatment
|
|
Case IIa
|
|
Plants not present at the time of treatment.
|
|
Case IIa Harvested crop not formed or barely formed
|
|
(e.g. applications — post-emergence
|
|
— post-planting
|
|
— applications pre-flowering)
|
|
Crop/group of crops
|
Number of trials
|
Comments
|
|
Citrus fruits
|
6
|
see Expl. 2
|
|
Tree nuts
|
6
|
see Expl. 2
|
|
Pome fruit
|
6
|
see Expl. 2
|
|
Stone fruit
|
6
|
see Expl. 2
|
|
Berries and small fruit:
|
|
|
|
( a ) Table and wine grapes
|
6
|
see Expl. 2
|
|
( b ) Strawberries
|
6
|
|
|
( c ) Cane fruits
|
6
|
see Expl. 2
|
|
( d ) Other small fruits and berries
|
6
|
see Expl. 2
|
|
( e ) Wild fruits
|
|
studies on ( b ), (c) and (d) are transferable to ( e )
|
|
Miscellanous fruits
|
8
|
see Expl. 1
|
|
|
|
The trials are to be spread over tree fruits and other miscellaneous fruits
|
|
Root and tuber vegetables
|
8
|
see Expl. 1
|
|
Bulb vegetables
|
6
|
see Expl. 2
|
|
Fruiting vegetables
|
12
|
see Expl. 3
|
|
Brassica vegetables
|
12
|
see Expl. 3
|
|
Leafy vegetables and fresh herbs
|
12
|
see Expl. 3
|
|
Legume vegetables (fresh) and pulses
|
6
|
see Expl. 2
|
|
Stem vegetables
|
8
|
see Expl. 1
|
|
Fungi including wild mushrooms
|
8
|
see Expl. 1
|
|
|
|
The studies are to be spread over soil-living and wood attacking fungi
|
|
Oil seeds
|
8
|
see Expl. 1
|
|
Potatoes
|
6
|
|
|
Tea
|
6
|
|
|
Hops
|
6
|
|
|
Cereals:
|
|
|
|
|
( a ) Barley
Oats
Rye
Wheat
Triticale
Buckwheat
|
|
6
|
i.e. studies on a minimum of one crop from each of Group (a) and (b) are required for the setting of a group tolerance for cereals
|
|
( b ) Maize including
sweet corn
Millet
Sorghum
Rice
|
|
6
|
|
|
Case IIb
|
|
Harvested crop formed
Case IIb 1.
Harvested crop grows totally or largely underground
|
|
Crop/group of crops
|
Number of trials
|
Comments
|
|
( a ) Fodder beet
Sugar beet
Swede/turnip
Beetroot
Celeriac
Radish
|
|
12
|
i.e. studies on a minimum of 2 crops (one major crop and one minor crop) are required for the setting of a tolerance for call the crops of the sub-group
|
|
( b ) Onions
Garlic
Shallots
|
|
6
|
i.e. studies on a minimum of one crop are required for the setting of a tolerance for all the crops of the sub-group
|
|
( c ) Carrots
Parsnips
Radish
Horseadish
Salsify
Asparagus
Potatoes
Turnip-rooted parsley
|
|
12
|
i.e. studies on a minimum of 2 crops (one major crop and one minor crop) are required for the setting of a tolerance for call the crops of the sub-group
|
|
Other crops
|
|
|
not relevant
|
|
Explanatory notes:
|
|
If studies are available showing comparable residue results for a minimum of 4 crops from the 3 sub-groups, then the setting of a maximum residue limit for all the crops named above can be discussed.
|
|
Case IIb 2. Harvested crop grows above the ground, but owing to the method of application comes into little or no contact with the product
|
|
(e.g. — applications of herbicides in crops growing high off the ground(fruit, vines, hops)
|
|
— treatments between the rows
|
|
— under-leaf treatments
|
|
|
|
Crop/group of crops
|
Number of trials
|
Comments
|
|
Citrus fruits
Tree nuts
Pome fruit
Stone fruit
|
|
12
|
see Expl. 3
|
|
Strawberries
|
6
|
|
|
Table and wine grapes
Cane fruits
Cane fruits
Other small fruits and berries
Wild fruits
|
|
12
|
see Expl. 3
|
|
Miscellaneous fruits:
Tree fruit
Other miscellaneous fruit
|
|
12
|
see Expl. 3
|
|
| | |
Crop/group of crops
|
Number of trials
|
Comments
|
|
Root and tuber vegetables
|
|
not relevant
|
|
Bulb vegetables
|
|
not relevant
|
|
Fruiting vegetables
|
12
|
see Expl. 3
|
|
Brassica vegetables
|
12
|
see Expl. 3
|
|
Leafy vegetables
|
12
|
see Expl. 3
|
|
Legume vegetables (fresh) and pulses
|
6
|
see Expl. 2
|
|
Stem vegetables
|
8
|
see Expl. 1
|
|
Fungi
|
|
not relevant
|
|
Oil seeds
|
8
|
see Expl. 1
|
|
Tea
|
6
|
|
|
Hops
|
6
|
|
|
Maize, including sweet corn
|
6
|
|
|
| | |
5.4.4
|
Special case
|
|
|
Rotational crops (including change of crop due to premature loss of the treated crop)
|
|
| |
Annex 1
|
| |
General Guidelines for Supervised Trials Programmes relating to Residues33
|
| |
1. The objectives of these studies are:
|
| |
(i) to establish the highest likely residue levels in treated crops at harvest or outloading from store; and
|
| |
(ii) to determine the rate of decline of pesticide deposits.
|
| |
2. MRL-Proposals are mainly based on results of supervised trials providing for worst-case test conditions — test conditions which in predictable circumstances will accommodate even the highest residues which may reasonably arise (maximum number of intentional applications, use of the maximum envisaged quantity) but which remain representative of conditions encountered in practice.
|
| |
3. MRLs are legal limits that are used for enforcement purposes, for the initial theoretical estimation of consumer exposure to pesticide residues and for facilitating trade. Hence, accuracy in their estimation is important, since Member States face very real difficulties when residues above the MRLs are determined. Factors influencing residue variability in crops should be taken into account when establishing a supervised trial programme. These would normally include for instance, climatic differences existing between production areas, differences in production methods, seasons of production, formulations etc. Data packages submitted which do not adequately take into account all possible sources of variability without satisfactory justification must be appropriately supplemented before a Community MRL will be further considered.
|
| |
4. Trials, in addition to being properly designed, executed and reported, must include treatments which correspond to GAP in the regions of authorization and use (e.g. minimum PHI, maximum rate and number of treatments, type of formulation).
|
| |
5. In general, for a comparable set of conditions, trials should be carried out over a minimum of two calendar years for crops with a single relatively narrow harvest period, e.g. grapes, apples. For products with an extended growing and harvesting season, such as, glasshouse lettuce, citrus fruit, sufficient data from two different growing seasons will normally do provided the GAP in question is the same.
|
| |
6. It is impossible and could be misleading to indicate the precise number of trials necessary to establish an MRL. However, experience indicates a minimum number of trials, below which the establishment of a MRL cannot be normally be considered. Minimum data requirements only apply where comparability can be established between production areas, e.g. concerning climate, methods and growing seasons of production etc Assuming all other variables (climate etc.) are comparable, a minimum of eight trials representative of the proposed growing area is required to establish a MRL for a major crop. For small geographical production areas, the number of trial locations may be reduced and replications conducted on fewer sites instead. For instance, instead of eight separately located trials, six trials with two replications per treatment or four trials with four replications per treatment may be acceptable. However, as variability mainly occurs between locations rather than within locations, the reduced number of trials must be truly representative of the production areas and the results show sufficient homogeneity between locations.
|
| |
33 The text comprises an extract from Commission Document 2946/VI/93: Study, prepared By Dr Jörg-Rainer Lundehn for the Commission of the European Communities, Guidelines for the Establishment of Community Maximum Residue Levels (MRLs) of Plant Protection Products in Food and Feedstuffs of Plant and Animal Origin, January 1993.
|
| |
7. Due to the inherently higher level of homogeneity in residues arising from post-harvest treatments, trials from one year (growing season) will be acceptable. In the case of fruit and vegetables, a minimum of four trials per application method (spray, dip, wax, etc.) and store type, carried out preferably on different locations and varieties, should normally be sufficient.
|
| |
In the case of post-harvest cereal, oil seed and pulse treatments, four trials with adequate sub-sampling to take account of residue variability within the store (e.g. vertical stratification) will normally be satisfactory for each application method.
|
| |
Trials from one year (growing seasons) will also be acceptable for some protected crops e.g. mushrooms and witloof because residue levels are not affected by variation of weather conditions.
|
| |
8. A certain percentage of the supervised residue trials reported, must include data to show the effect of time on the level of residue present (test series) where:
|
| |
(i) it is clear that residues are likely to occur at or close to harvest;
|
| |
(ii) if the plant protection product is to be applied when the edible portion or the portion to be used, has formed; or
|
| |
(iii) it is clear that residues may occur at, or close to, the earliest harvest time, and harvesting can or does take place over an extended period e.g. root crops.
|
| |
9. In test series the number of points and the intervals between them will be influenced by the half-life of the chemical and the anticipated pre-harvest interval. However, curves should normally have five points with a minimum of three, initial deposit and deposit on the day of harvest should always be included. When designing trials, due consideration should be given to possible variation and changes in GAP. Inclusion of a number well chosen pre-harvest intervals may avoid the necessity of repeating trials following changes in GAP.
|
| |
10. Whilst full unabridged data should be available to the rapporteur, data on GAP and supervised trials should be summarized on the relevant summary reporting forms.
|
| |
11. Detailed guidelines relating to the design, conducting and reporting of residue trials are available, on request, from the competent authority.
|
| |
Annex 2
|
| |
List of major crops
|
| | |
Crops not mentioned are assumed to be minor crops
|
|
N = Northern Region (Europe)
|
|
S = Southern and Mediterranean Region (Europe)
|
|
W = World (except Europe)
|
|
| | |
|
|
N
|
S
|
W
|
|
Citrus fruit
|
|
|
|
|
|
|
Oranges
|
|
X
|
X
|
|
|
Mandarins/clementines/satsumas
|
|
X
|
X
|
|
|
Lemons
|
|
X
|
X
|
|
|
Grapefruit/pomelo
|
|
|
X
|
|
Tree nuts
|
|
|
|
|
|
|
Almonds
|
|
X
|
X
|
|
|
Pistachios
|
|
|
X
|
|
|
Hazelnuts
|
|
|
X
|
|
|
Cashew nuts
|
|
|
X
|
|
|
Chestnuts
|
|
|
X
|
|
|
Walnuts
|
X
|
X
|
X
|
|
|
Pecans
|
|
|
X
|
|
|
Coconuts
|
|
|
X
|
|
Pome fruit
|
|
|
|
|
|
|
Apples
|
X
|
X
|
X
|
|
|
Pears
|
X
|
X
|
X
|
|
Stone fruit
|
|
|
|
|
|
|
Peaches/nectarines
|
|
X
|
X
|
|
|
Plums
|
X
|
X
|
X
|
|
|
Apricots
|
|
X
|
X
|
|
|
Cherries
|
X
|
X
|
X
|
|
Grapes
|
|
|
|
|
|
|
Wine grapes
|
X
|
X
|
X
|
|
|
Table grapes
|
|
X
|
X
|
|
Berries and small fruit
|
|
|
|
|
|
|
Strawberries
|
X
|
X
|
X
|
|
|
Raspberries
|
X
|
|
X
|
|
|
Currants
|
X
|
|
|
|
|
|
|
|
|
|
Miscellaneous fruits
|
|
|
|
|
|
|
Dates
|
|
|
X
|
|
|
Avocados
|
|
|
X
|
|
|
Mangoes
|
|
|
X
|
|
|
Pineapples
|
|
|
X
|
|
|
Bananas
|
|
X
|
X
|
|
|
Papayas
|
|
|
X
|
|
|
Kiwi fruit
|
|
X
|
X
|
|
|
Olives (oil production)
|
|
X
|
X
|
|
Root and tuber vegetables
|
|
|
|
|
|
|
Potatoes
|
X
|
X
|
X
|
|
|
Sweet potatoes
|
|
|
X
|
|
|
Cassava
|
|
|
X
|
|
|
Yams
|
|
|
X
|
|
|
Taro (coco yam)
|
|
|
X
|
|
|
Carrots
|
X
|
X
|
X
|
|
|
Sugar beet
|
X
|
X
|
X
|
|
|
Fodder beet
|
X
|
|
|
|
|
|
|
|
|
|
|
Turnips/swedes
|
X
|
|
X
|
|
Bulb vegetables
|
|
|
|
|
|
|
Onions
|
X
|
X
|
X
|
|
|
Garlic
|
|
X
|
X
|
|
Fruiting vegetables
|
|
|
|
|
|
|
Tomatoes
|
X
|
X
|
X
|
|
|
Cucumbers and gherkins
|
X
|
X
|
X
|
|
|
Eggplants
|
X
|
X
|
X
|
|
|
Peppers
|
|
X
|
X
|
|
|
Watermelons
|
|
X
|
X
|
|
|
Melons
|
|
X
|
X
|
|
|
Sweet corn
|
|
|
X
|
|
Brassica vegetables
|
|
|
|
|
|
|
Cabbages, head
|
X
|
X
|
X
|
|
|
Cauliflower
|
X
|
X
|
X
|
|
|
Brussels sprouts
|
X
|
|
|
|
|
Kale
|
X
|
|
|
|
|
Chinese cabbage
|
|
X
|
X
|
|
Leafy vegetables
|
|
|
|
|
|
|
Lettuce, head
|
X
|
X
|
X
|
|
|
Spinach
|
X
|
X
|
X
|
|
|
Witloof/chicory
|
X
|
|
X
|
|
|
Endive/scarole
|
X
|
X
|
X
|
|
Legume vegetables (fresh)
|
|
|
|
|
|
|
Beans, green
|
X
|
X
|
X
|
|
|
Peas, green
|
X
|
X
|
X
|
|
Stem vegetables
|
|
|
|
|
|
|
Artichokes, globe
|
X
|
X
|
|
|
|
Leeks
|
X
|
X
|
X
|
|
Pulses
|
|
|
|
|
|
|
Beans, dry
|
X
|
X
|
X
|
|
|
Peas, dry
|
X
|
X
|
X
|
|
|
Chick-peas
|
|
X
|
X
|
|
|
Lentils
|
|
X
|
X
|
|
Oil seed
|
|
|
|
|
|
|
Soya bean
|
|
X
|
X
|
|
|
Peanuts
|
|
|
X
|
|
|
Sunflower seed
|
X
|
X
|
X
|
|
|
Rapeseed
|
X
|
X
|
X
|
|
|
Sesame seed
|
|
|
X
|
|
|
Linseed
|
X
|
|
X
|
|
|
Safflower seed
|
|
|
X
|
|
|
Cotton seed
|
|
X
|
X
|
|
|
Copra
|
|
|
X
|
|
|
Palm kernels
|
|
|
X
|
|
Cereals
|
|
|
|
|
|
|
Wheat/triticale
|
X
|
X
|
X
|
|
|
Rice, paddy
|
|
X
|
X
|
|
|
Barley
|
X
|
X
|
X
|
|
|
Maize
|
X
|
X
|
X
|
|
|
Rye
|
X
|
X
|
X
|
|
|
Oats
|
X
|
X
|
X
|
|
|
Millet
|
|
|
X
|
|
|
Sorghum
|
|
X
|
X
|
|
Miscellaneous
|
|
|
|
|
|
|
Sugar cane
|
|
|
X
|
|
|
Coffee, green
|
|
|
X
|
|
|
Cocoa beans
|
|
|
X
|
|
|
Tea
|
|
|
X
|
|
|
Hops
|
X
|
|
X
|
|
|
Tobacco leaves
|
|
X
|
X
|
|
|
Mushrooms (except wild mushrooms)
|
X
|
X
|
X
|
|
| |
SIXTH SCHEDULE
|
| |
PART 1
|
| |
Regulations 19 (1)
|
| |

|
| |

|
| |

|
| |
PART 2
|
| |
Regulations 19 (2)
|
| |

|
| |

|
| |

|
| |
PART 3
|
| |
Regulations 19 (4)
|
| |

|
| |

|
| |

|
| |
PART 4
|
| |
Regulations 19 (5)
|
| |

|
| |

|
| |
SEVENTH SCHEDULE
|
| |
Regulations 25 (1) and 26 (4)
|
| |
Principles of Good Experimental Practice
|
| | |
1
|
Introduction
|
|
|
The prime purpose of the Principles of Good Experimental Practice (GEP) is to promote the development of quality test data. A further objective is to facilitate the mutual acceptance of test data for regulatory purposes by other Member States of the Community, thereby avoiding duplicative testing and achieving economies in test costs and time, where test conditions are shown to be comparable. The Principles of Good Experimental Practice are analogous to the Principles of Good Laboratory Practice (GLP). They are designed to facilitate the production of quality data, generated through testing at many sites, including those which are not directly controlled by those conducting tests, without imposing impractical quality assurance and inspection obligations.
|
|
|
These Principles of Good Experimental Practice apply to the testing of the performance (efficacy and phytotoxicity) of plant protection products, including adjuvants, in support of applications for the authorization of plant protection products. These Principles also apply to the testing of both active substances and plant protection products, with respect to physical and chemical properties and technical characteristics, where the tests do not relate to their properties or safety with respect to human health or the environment. Testing with respect to effects on honey bees and beneficial arthropods other then bees (paragraph 2.2 of the introduction to Annex II, paragraph and 2.3, Annex III, points 2.2 and 2.3), in accordance with these Principles of Good Experimental Practice, as opposed to GLP, will be accepted if initiated prior to 31 December 1999 (see point 2.2 of the introduction to Annex II and point 2.4 of the introduction to Annex III).
|
|
2
|
Definitions
|
|
2.1
|
testing facility:
|
|
|
The persons, premises, and operational unit(s) necessary for testing. The term may include several sites, at one or more geographical locations, where phases or components of a single overall programme of tests are conducted. The different sites may include, but are not limited to:
|
|
|
— laboratory(ies) where the test or other material is characterized, measured, or otherwise assessed;
|
|
|
— one or more agricultural or other in- or outdoor sites, including glasshouses, where the test material is applied to the test system; and
|
|
|
— where relevant a processing facility where harvested commodities are treated to prepare other items, e.g. conversion to sugar, flour, beer, puree, juice.
|
|
2.2
|
test:
|
|
|
Technical operation that consists of the determination of one or more characteristics or effects of a given active substance, plant protection product, or adjuvant, according to a specified procedure.
|
|
3
|
Legal Identity
|
|
|
The testing facility shall be legally identifiable.
|
|
4
|
Impartiality, independence and integrity
|
|
|
The testing facility and its personnel shall be free from any commercial, financial and other pressures which might influence their technical judgement.
|
|
|
Any influence on the results of examinations and tests exercised by persons or organizations external to the testing facility shall be excluded.
|
|
|
The testing facility shall not engage in any activities that may endanger the trust in its independence of judgement and integrity in relation to its testing activities.
|
|
|
When products are tested by bodies, who have been or are concerned with their design, development, manufacture or sale, provision shall be made for a clear separation of the different responsibilities and for an appropriate statement as to responsibilities.
|
|
5
|
Technical competence
|
|
5.1
|
Management and organization
|
|
|
The testing facility shall be competent to perform the tests concerned. In the absence of a recognized test guideline, agreement between the client and the testing facility to the test procedure shall be documented.
|
|
|
The testing facility shall be organized in such a way that each member of personnel is aware of both the extent and the limitation of his area of responsibility.
|
|
|
The organization shall provide supervision by persons familiar with relevant test methods and procedures, the objective of such testing and the assessment of test results. The proportion of supervisory to non-supervisory personnel shall be such as to ensure adequate supervision.
|
|
|
The testing facility shall have a technical manager who has overall responsibility for the technical operations of the facility.
|
|
|
A document showing the organization and distribution of responsibilities of the testing facility shall be available and kept up-to-date.
|
|
5.2
|
Personnel
|
|
|
The testing facility shall have sufficient personnel, having the necessary education, training, technical knowledge and experience for their assigned functions.
|
|
|
The testing facility shall ensure that the training of its personnel is kept up-to-date.
|
|
|
Information on the relevant qualifications, training and experience of the technical personnel shall be maintained by the facility.
|
|
5.3
|
Premises and equipment
|
|
5.3.1
|
Availability
|
|
|
The testing facility shall be furnished with all items of equipment required for correct performance of the tests and measurements which it claims to be competent to carry out.
|
|
|
In the exceptional case where the facility is obliged to use outside equipment, it shall ensure the quality of that equipment.
|
|
5.3.2
|
Premises and environment
|
|
|
The environment in which the tests are undertaken shall not invalidate the test results or adversely affect the required accuracy of measurement. The premises shall have the equipment, energy sources and other services needed for the testing. When the testing so requires, they shall be equipped with devices to monitor environmental conditions.
|
|
|
Access to and use of all test areas shall be controlled in a manner appropriate to their designated purpose and conditions of entry by persons external to test sites shall be defined.
|
|
|
In the case of premises used by a testing facility which are not subject to the exclusive control of the testing facility, arrangements shall be made to control access to and use of test areas in a manner appropriate to their designated purpose. The conditions of entry by persons external to the testing facility and the means of ensuring compliance with those conditions shall be defined.
|
|
|
Adequate measures shall be taken to ensure good house-keeping in the testing facility.
|
|
5.3.3
|
Equipment
|
|
|
All equipment shall be properly maintained. Details of maintenance procedures shall be available.
|
|
|
Any item of the equipment which has been subjected to overloading or mishandling, or which gives suspect results, or has been shown by calibration or otherwise to be defective, shall be taken out of service, clearly labelled and stored at a specified place until it has been repaired and then shown by test or calibration to be performing its function satisfactorily. The testing facility shall examine the effect of this defect on previous tests and report its findings to the competent authority.
|
|
|
Records shall be maintained of each major item equipment used in testing. Each record shall include:
|
|
|
( a ) the name of the item of equipment;
|
|
|
( b ) the manufacturer's name and type identification and serial number;
|
|
|
( c ) date received and date placed in service;
|
|
|
( d ) current location, where appropriate;
|
|
|
( e ) condition when received (e.g. new, used, reconditioned)
|
|
|
( f ) details of maintenance carried out; and
|
|
|
( g ) history of any damage, malfunction, modification or repair.
|
|
|
Measuring, testing and dispensing (including application) equipment used in the testing facility shall be calibrated where appropriate before being put into service and thereafter according to an established programme.
|
|
|
The overall programme of calibration of equipment shall be designed and operated so as to ensure that wherever applicable measurements made in the testing facility are traceable to national and international standards of measurement. Where traceability to national or international standards of measurement is not applicable, the testing facility shall provide satisfactory evidence of correlation or accuracy of test results (for example by participation in a suitable programme of inter laboratory comparisons).
|
|
|
Reference standards of measurement held by the testing facility shall be used for calibration only and for no other purpose.
|
|
|
Reference standards of measurement shall be calibrated by a competent body that can provide traceability to a national or international standard of measurement.
|
|
|
Where relevant, testing equipment shall be subjected to in-service checks between regular re-calibrations. The calibration of application equipment shall be checked on each occasion on which it is used in a test.
|
|
|
Reference materials shall where possible be traceable to national or international standard reference materials.
|
|
5.4
|
Working procedures
|
|
5.4.1
|
Test methods and procedures
|
|
|
The testing facility shall have adequate documented instructions on the use and operation of all relevant equipment, on the handling and preparation of test materials, and on standard testing techniques. All instructions, standards, manuals and reference data relevant to the work of the testing facility shall be maintained up-to-date and be readily available to the personnel.
|
|
|
The testing facility shall use methods and procedures required by or acceptable to the competent authority for the testing of plant protection products. The experimental protocol to be followed shall be provided to personnel performing the test, together with all relevant documented instructions on the handling and preparation of test materials, and on standard testing techniques.
|
|
|
The testing facility shall reject requests to perform tests according to test methods that may endanger an objective result, have a reduced validity or are unacceptable to the competent authority.
|
|
|
Where it is necessary to employ test methods and procedures which are non-standard, these shall be fully documented.
|
|
|
All calculation and data transfer shall be subject to appropriate checks.
|
|
|
Where results are derived by electronic data processing techniques, the reliability and stability of the system shall be such that the accuracy of the results is not affected. The system shall be able to detect malfunctions during programme execution, to permit appropriate action to be taken.
|
|
5.4.2
|
Quality System
|
|
|
The testing facility shall operate a Quality System appropriate to the type, range and volume and intended purpose of work performed. The elements of this system shall be documented in a Quality Manual which is available for use by the personnel responsible for and involved in testing. The Quality Manual shall be maintained current by a nominated responsible member of the testing facility personnel.
|
|
|
A person or persons having responsibility for quality assurance within the facility shall be designated by the facility management and have direct access to top management.
|
|
|
The Quality Manual shall contain at least:
|
|
|
( a ) a quality policy statement;
|
|
|
( b ) the structure of the laboratory (organizational charts);
|
|
|
( c ) the operational and functional activities pertaining to quality, so that each person concerned will know the extent and the limits of his responsibility;
|
|
|
( d ) general quality assurance procedures;
|
|
|
( e ) reference to quality assurance procedures specific for each test, as appropriate;
|
|
|
( f ) where appropriate, reference to proficiency testing, use of reference material, etc. ;
|
|
|
( g ) satisfactory arrangements for feedback and corrective action whenever testing discrepancies are detected; and
|
|
|
( h ) procedure for dealing with complaints.
|
|
|
The quality system shall be systemically and periodically reviewed by or on behalf of management to ensure the continued effectiveness of the arrangements, and any necessary corrective action initiated. Such reviews shall be recorded together with details of any corrective action taken.
|
|
5.4.3
|
Test reports
|
|
|
The work carried out by the testing facility shall be covered by a report which accurately, clearly and unambiguously presents the test results and all other relevant information.
|
|
|
In addition to the information to be reported as specified in relevant test guidelines, each test report shall include at least the following information:
|
|
|
( a ) name and address of testing facility and the precise location where the test was carried out;
|
|
|
( b ) unique identification of the report (such as serial number) and of each page, and total number of pages of the report;
|
|
|
( c ) name and address of client;
|
|
|
( d ) description and identification of the test material;
|
|
|
( e ) date of receipt of test material and date(s) of performance of test;
|
|
|
( f ) identification of the test guideline or description of the method;
|
|
|
( g ) description of sampling procedure, where relevant;
|
|
|
( h ) any deviations, additions to or exclusions from the test guideline, and any other information relevant to a specific test;
|
|
|
( i ) identification of any non-standard test method or procedure utilized, and a full description of the test method;
|
|
|
( j ) measurements, examinations and derived results, supported by tables, graphs, sketches and photographs as appropriate;
|
|
|
( k ) a statement on measurement uncertainty (where relevant);
|
|
|
( l ) a signature and title or an equivalent marking of person(s) accepting technical responsibility for the test report and date of issue; and
|
|
|
( m ) a statement to the effect that the test results relate only to the items tested.
|
|
|
Particular care and attention shall be paid to the arrangement of the test report, especially with regard to presentation of the test data and ease of assimilation by the reader. The format shall be carefully and specifically designed for each type of test carried out, but the headings shall be standardized as far as possible.
|
|
|
Corrections or additions to a test report after issue shall be made only by a further document suitably marked, e.g.
|
|
NOTE:
|
|
Test results could be measured values, findings from the visual examination or practical use of the test material, derived results or any other type of observation from the testing activities. Test results could be supported by tables, photographs, or graphical information of any kind, appropriately identified.
|
|
|
"Amendment/Addendum to test report serial number ...(or as otherwise identified)", and shall meet the relevant requirements of the preceding paragraphs.
|
|
|
A test report shall not include any advice or recommendation arising from the test results.
|
|
|
Test results shall be presented accurately, clearly, completely and unambiguously in accordance with instructions that may be part of the test methods.
|
|
|
Quantitative results shall be given together with calculated or estimated uncertainty.
|
|
5.4.4
|
Records
|
|
|
The testing facility shall maintain a record system to suit its particular circumstances and comply with any existing regulations. It shall retain on record all original observations, calculations and derived data, calibration records and the final test report for as long as the substance or product concerned continues to be authorized or placed on the market in the Community. The records for each test shall contain sufficient information to permit repetition of the test. The records shall include the identity of personnel involved in calibration of equipment, sampling, and reading of measurements.
|
|
|
All records and test reports shall be safely stored, held secure and in confidence to the client and the competent authority, unless otherwise required by law.
|
|
5.4.5
|
Handling of test samples or items
|
|
|
A system for identifying the samples or materials to be tested shall be applied, either through documents or through marking, to ensure that there can be no confusion regarding the identity of the samples or items and the results of the measurements made.
|
|
|
The system shall include arrangements to ensure that the samples or items can be handled anonymously, e.g. to other clients.
|
|
|
A procedure shall exist for bonded storage of samples or items where necessary.
|
|
|
At all stages of storing, handling and preparation for test, precautions shall be taken to prevent damage to the samples or test material, for example contamination or the application of stresses (temperature), which would invalidate the results of testing. Any relevant instructions provided with the samples or materials to be tested shall be observed.
|
|
|
There shall be clear rules for the receipt, retention and disposal of samples and test materials.
|
|
5.4.6
|
Confidentiality and security
|
|
|
The personnel of the testing facility shall be bound to observe professional secrecy with regard to all information gained in carrying out its tasks.
|
|
|
The testing facility shall observe terms and conditions to provide for confidentiality and security of its practices as required by the user of its services.
|
|
5.4.7
|
Subcontracting
|
|
|
Testing facilities shall themselves normally perform the testing which they contract to undertake. Where, exceptionally, a testing facility subcontracts any part of the testing, this work shall be placed with another testing facility complying with these requirements. The testing facility shall ensure and be able to demonstrate that its subcontractor is competent to perform the services in question and complies with the same criteria of competence as the testing facility in respect of the work being subcontracted. The testing facility shall advise the client of its intention to subcontract any portion of the testing to another party. The subcontractor shall be acceptable to the client.
|
|
|
The testing facility shall record and retain details of its investigation of the competence and the compliance with the requirements of these Principles of GEP of its subcontractors and maintain a register of all subcontracting.
|
|
6
|
Cooperation
|
|
6.1
|
Cooperation with clients
|
|
|
The testing laboratory shall afford the client or his representative cooperation to enable him to clarify the client's request and to monitor the performance of the testing facility in relation to the work to be performed. This cooperation shall include:
|
|
|
( a ) affording the client or his representative access to relevant areas of the testing facility, for the witnessing of tests performed for the client. It is understood that such access shall by no means come into conflict with rules of confidentiality of work for other clients and with safety; and
|
|
|
( b ) preparation, packaging and dispatch of test samples or materials needed by the client for verification purposes.
|
|
|
The testing facility shall have a defined complaint procedure. This shall be documented and made available on request.
|
|
6.2
|
Cooperation with competent authority
|
|
|
The testing facility shall afford the competent authority and its authorized officers such reasonable cooperation as is necessary, to enable the competent authority to monitor compliance with these requirements and other relevant obligations. This cooperation shall include:
|
|
|
( a ) affording authorized officers access to relevant areas of the testing facility, for the witnessing of tests;
|
|
|
( b ) undertaking any reasonable checks to enable the competent authority to verify the testing capability of the testing facility;
|
|
|
( c ) providing access to and copies of all relevant documentation and records necessary to verify compliance with these requirements;
|
|
|
( d ) preparation, packaging and dispatch of test samples or items needed by the competent authority for verification purposes;
|
|
|
( e ) participation in any appropriate programme of proficiency testing or comparison testing that the competent authority may reasonably deem to be necessary;
|
|
|
( f ) permitting scrutiny by the competent authority of the results of the testing facility's own internal audits and where appropriate, proficiency tests; and
|
|
|
( g ) where requested, making available, prior to the commencement of a test, detailed information concerning it, including its location and the materials to be tested.
|
|
6.3
|
Cooperation with other laboratories and with bodies producing standards and guidelines
|
|
|
Testing facilities are encouraged to participate in the drawing up of national, European or international standards and guidelines in the area of testing, where appropriate.
|
|
|
Testing facilities are encouraged to take part in an exchange of information with other laboratories and facilities having test activities in the same technical field where appropriate. The object is to have uniform test procedures and guidelines and to improve the quality of testing where appropriate.
|
|
|
In order to maintain the accuracy required, regular comparison of test results by proficiency testing should be arranged where appropriate.
|
|
7
|
Further duties of testing facilities
|
|
|
Testing facilities shall:
|
|
|
( a ) before undertaking testing, ensure that necessary authorizations for trials purposes have been issued by the competent authority, unless the testing concerning is to be conducted in accordance with the terms and conditions of a valid trials permit;
|
|
|
( b ) ensure, where relevant, that testing is conducted in accordance with the conditions of the relevant authorization for trials purposes;
|
|
|
( c ) at all times comply with the requirements of these Principles of GEP and with other criteria prescribed by the competent authority;
|
|
|
( d ) an annual report to the competent authority, containing all detailed information necessary to demonstrate compliance with these Principles of GEP;
|
|
|
( e ) claim that the Principles of Good Efficacy Testing Practice have been complied with only in respect of testing services which are carried out in accordance with the requirements of this Schedule and other criteria prescribed by the competent authority;
|
|
|
( f ) not use its compliance with this Schedule in such a manner as to bring the competent authority into disrepute and not make any statement relevant to compliance or non-compliance which the competent authority may reasonably consider to be misleading;
|
|
|
( g ) make it clear in all contracts with its clients that compliance with the Principles of Good Efficacy Testing Practice or test reports generated in accordance with those principles, by themselves in no way constitute or imply product authorization or approval by the competent authority or any other body;
|
|
|
( h ) ensure that no test report nor any part thereof shall be used by them, or be authorized for use by them, for promotional or publicity purposes, if the competent authority considers such use to be misleading; and
|
|
|
( i ) inform immediately the competent authority of any changes bearing on its compliance with the requirements of this Schedule and other criteria affecting the testing facility's capability or scope of activity.
|
|
| |
EIGHT SCHEDULE
|
| |
PART 1
|
| |
Regulations 25 (3)
|
| |

|
| |

|
| |

|
| |

|
| |
|
| |
|
| |
|
| |
|
| |

|
| |

|
| |

|
| |
|
| |

|
| |

|
| |

|
| |
PART 2
|
| |
Regulations 25 (12)
|
| |

|
| |

|
| |
PART 3
|
| |
Regulations 25 (12)
|
| |

|
| |

|
| |
NINTH SCHEDULE
|
| |
Regulations 25 (5)
|
| |
DATA AND INFORMATION REQUIRED IN SUPPORT OF APPLICATIONS FOR AUTHORIZATIONS FOR TRIALS PURPOSES
|
| |
INTRODUCTION
|
| | |
1
|
Although the extent of the data and information required to support applications for the authorization of plant protection products for trials purposes is quite limited, the tests and studies reported must satisfy the same standards as to quality, as is required of tests and studies submitted in support of applications for authorization for commercial marketing and use.
|
|
2
|
The information submitted must:
|
|
2.1
|
include a technical dossier supplying the information necessary for evaluating the foreseeable immediate risks which the plant protection product may entail for humans, animals and the environment, as well as the information necessary for the identification of potential delayed risks for consumers through dietary exposure, and contain at least the information and results of the studies referred to below;
|
|
2.2
|
where relevant, be generated using test guidelines referred to or described in Annex II or Annex III, or in this Schedule, in the case of studies initiated before the adoption of the modification of those Annexes, or before these Regulations come into effect, as appropriate, the information shall be generated using suitable internationally validated test guidelines or, in the absence thereof, test guidelines accepted by the competent authority;
|
|
2.3
|
in the event of a test guideline being inappropriate or not described, or where another one than those referred to in annex II or III or in this Schedule has been used, include a justification, which is acceptable to the competent authority for the guideline used;
|
|
2.4
|
include, a full description of test guidelines used, except if they are referred to or described in Annex II or III, or in this Schedule, and a full description of any deviations from the guidelines used, including a justification, which is acceptable to the competent authority, for these deviations;
|
|
2.5
|
include a full and unbiased report of the studies conducted as well as a full description of them, or a justification which is acceptable to the competent authority, where:
|
|
|
— particular data and information which would not be necessary owing to the nature of the product or its proposed uses, are not provided, or
|
|
|
— it is not scientifically necessary, or technically possible to supply information and data; and
|
|
2.6
|
where relevant, have been generated in accordance with the requirements of Directive 86/609/EEC.
|
|
3
|
Tests and analyses must be conducted in accordance with the principles laid down in Directive 87/18/EEC, where testing is done to obtain data on the properties and/or safety with respect to human or animal health or the environment.
|
|
4
|
The information required must include the proposed classification and labelling of the plant protection product in accordance with relevant Community Directives, as well as the proposed classification and labelling of each novel active substances contained in the plant protection product.
|
|
5
|
Individual study reports must be presented in accordance with the requirements of Part 2 of the Fourth Schedule. Subject to the appropriate clearance or authorization number being quoted, study reports already submitted for plant protection products cleared in accordance with the Regulations of 1994, or authorized in accordance with Regulations 13, 15, or 18, need not be submitted again in support of applications for authorization for trials purposes.
|
|
6
|
The information required must include a summary and overview of the information and study reports submitted. Where particular information, test reports or studies specified are not provided, or have not been submitted previously, the omission must be justified and a reasoned case must be made justifying the granting of an authorization for trials purposes, in the absence of the data concerned. Additional data, not specified hereunder, can be provided in support of applications, where it is of suitable quality, and relates to the risks which the plant protection product may entail for humans, animals and the environment.
|
|
7
|
Authorizations for trials purposes cannot be granted where there are significant gaps in the supporting documentation provided. Decisions as to whether an authorization for trials purposes can be granted where there are minor gaps in the supporting documentation provided, can only be made on a case by case basis. The following general guidance is provided:
|
|
7.1
|
in all cases at least that information and data concerning the identity of each active substance, the physical and chemical properties of each active substance, the identity of the plant protection product, the physical and chemical properties of the plant protection product and the acute toxicity of each active substance, must be provided;
|
|
7.2
|
in the case of preparations, which classify as Very Toxic, or Toxic (on the basis of calculations), or which contain substances which are allergic sensitizers, data relating to the acute toxicity of the preparation, must be provided; and
|
|
7.3
|
where the nature of the proposed use is such that a there is a moderate or high risk of residues occurring in food or feed commodities, and where it is not proposed that produce be destroyed, data relating to the short-term toxicity and genotoxicty profile of each active substance, and data on residues at harvest, must be provided.
|
|
| |
PART A
|
| |
Chemical substances
|
| | |
|
Identity of the applicant
|
|
|
The name and address of the applicant (permanent Community address) must be provided as must the name, position, telephone and telefax number of the appropriate person to contact.
|
|
|
Where, in addition, the applicant has an office, agent or representative within the territory of the state, the name and address of the local office, agent or representative must be provided as must the name, position, telephone and telefax number of the appropriate person to contact.
|
|
2
|
Identity of the active substance(s)
|
|
|
The information provided must be sufficient, to identify with precision each active substance in the plant protection product, to define it in terms of its specification and to characterize it as to its nature. The information and data referred to, unless otherwise specified, are required for all active substances.
|
|
2.1
|
Common name proposed or ISO-accepted, and synonyms
|
|
|
For each active substance in the plant protection product, the ISO common name, or proposed ISO common name, and where relevant, other proposed or accepted common names (synonyms), including the name (title) of the nomenclature authority concerned, must be provided.
|
|
2.2
|
Chemical name (IUPAC and CA nomenclature)
|
|
|
For each active substance in the plant protection product, the chemical name as given in Annex I to the Directive of 1967, or if not included in that Directive, in accordance with both IUPAC and CA nomenclature, must be provided.
|
|
2.3
|
Manufacturer's development code number(s)
|
|
|
For each active substance in the plant protection product, code numbers used to identify the active substances concerned and, where available, to identify formulations containing each of the active substances during development work, must be reported. For each code number reported, the material to which it relates, the period for which it was used, and the Member States or other countries in which it was used, and is being used, must be reported.
|
|
2.4
|
CAS and EEC numbers (if available)
|
|
|
For each active substance in the plant protection product, the Chemical Abstracts, EEC (EINECS or ELINCS), and CIPAC numbers, where they exist, must be reported.
|
|
2.5
|
Molecular and structural formula, molecular mass
|
|
|
The molecular formula, molecular mass and structural formula of each active substance, and where relevant, the structural formula of each stereo and optical isomer present in the active substances concerned, must be provided.
|
|
2.6
|
Manufacturer (name, address, including location of plant)
|
|
|
The name and address of the manufacturer or manufacturers of each active substance must be provided as must the name and address of each manufacturing plant in which the active substances concerned are manufactured. A contact point (preferably a central contact point, to include name, telephone and telefax number) must be provided, with a view to providing updating information and responding to queries arising, regarding manufacturing technology, processes and the quality of technical product (including where relevant, individual batches). Where following authorization for trials purposes, there are changes in the location or number of manufacturers, the information required must again be provided, unless the period of validity of the relevant authorization for trials purposes has expired or it has not been renewed.
|
|
2.9
|
Method of manufacture (synthesis pathway) of active substance
|
|
|
For each active substance in the plant protection product, the method of manufacture, in terms of the identity of the starting materials, the chemical pathways involved, and the identity of by-products and impurities present in the final product, must be provided, for each manufacturing plant. Generally, process engineering information is not required.
|
|
|
Where the information provided relates to a laboratory or pilot plant production system, the information required must again be provided once industrial scale production methods and procedures have stabilized, unless the period of validity of the relevant authorization for trials purposes has expired or it has not been renewed.
|
|
2.10
|
Specification of purity of active substance in g/kg or g/l as appropriate
|
|
|
For each active substance in the plant protection product, the minimum content in g/kg of the pure active substance (excluding inactive isomers) in the manufactured material used in production of the formulated product, must be reported.
|
|
|
Where the information provided relates to a laboratory or pilot plant production system, the information required must again be provided once industrial scale production methods and procedures have stabilized, if production changes result in a changed specification of purity, unless the period of validity of the relevant authorization for trials purposes has expired or it has not been renewed.
|
|
2.11
|
Identity of isomers, impurities and additives (e.g. stabilizers), together with the structural formula and the content expressed as g/kg
|
|
|
For each active substance in the plant protection product, the maximum content in g/kg of inactive isomers as well as the ratio of the content of isomers/diastereoisomers, where relevant, must be provided. In addition, the maximum content in g/kg of each further component other than additives, including by-products and impurities must be provided. In the case of additives, the content in g/kg must be provided.
|
|
|
For each component, present in quantities of 1 g/kg or more, the following information, where relevant, must be provided:
|
|
|
— chemical name according to IUPAC and CA nomenclature;
|
|
|
— ISO common name or proposed common name, if available;
|
|
|
— CAS number, EEC (EINECS or ELINCS) number, and CIPAC number, if available;
|
|
|
— molecular and structural formula;
|
|
|
— molecular mass;
|
|
|
— optical purity and ratio of enantiomorphs; and
|
|
|
— maximum content in g/kg.
|
|
|
Where the manufacturing process is such that impurities and by-products which are particularly undesirable because of their toxicological or environmental properties could be present in an active substance, the content of each such compound must be determined and reported. In such cases, the analytical methods used and the limits of determination, which must be sufficiently low, for each compound of concern, must be reported. Additionally, the following information, where relevant, must be provided:
|
|
|
— chemical name according to IUPAC and CA nomenclature;
|
|
|
— ISO common name or proposed common name, if available;
|
|
|
— CAS number, EEC (EINECS or ELINCS) number, and CIPAC number, if available;
|
|
|
— molecular and structural formula;
|
|
|
— molecular mass;
|
|
|
— optical purity and ratio of enantiomorphs; and
|
|
|
— maximum content in g/kg.
|
|
|
Where the information provided does not fully identify a component viz. condensates, detailed information on their composition must be provided for each such component
|
|
|
Where the information provided relates to a laboratory or pilot plant production system, the information required must again be provided once industrial scale production methods and procedures have stabilized, if production changes result in a changed specification of purity, unless the period of validity of the relevant authorization for trials purposes has expired or it has not been renewed.
|
|
|
The trade name of components added to active substances, prior to manufacture of formulated product, to preserve stability and facilitate ease of handling, where they are used, must also be provided. Additionally, the following information, where relevant, must be provided for such additives:
|
|
|
— chemical name according to IUPAC and CA nomenclature;
|
|
|
— ISO common name or proposed common name, if available;
|
|
|
— CAS number, EEC (EINECS or ELINCS) number, and CIPAC number, if available;
|
|
|
— molecular and structural formula;
|
|
|
— molecular mass;
|
|
|
— optical purity and ratio of enantiomorphs; and
|
|
|
— maximum mean content in g/kg.
|
|
For added components, other than active substance and other than impurities resulting from the manufacturing process, the function of the component (additive) must be given:
|
|
antifoaming agent
|
buffer
|
other (specify)
|
|
antifreeze
|
dispersing agent
|
|
|
binder
|
stabilizer
|
|
|
3
|
Physical and chemical properties of the active substance(s)
|
|
(i)
|
The information provided, must describe the physical and chemical properties of each active substance included in the plant protection product and together with other relevant information, must serve to characterize them. In particular, the information provided must permit:
|
|
|
— physical, chemical, and technical hazards associated with active substances, to be identified;
|
|
|
— classification of active substance as to hazard;
|
|
|
— appropriate restrictions and conditions to be associated with any authorization for trials purposes granted to be selected; and
|
|
|
— appropriate risk and safety phrases to be specified.
|
|
|
The information and data referred to are required for all active substances, except where otherwise specified.
|
|
(ii)
|
The information provided, taken together with that provided for the formulated product, must permit the physical, chemical and technical hazards associated with preparations, to be identified, permit preparations to be classified, and establish that preparations can be used without unnecessary difficulty, and be such that exposure of man, animals, and the environment is minimized, taking account of manner of proposed use.
|
|
(iii)
|
Data and information as to the physical and chemical properties of active substances, are of particular value in predicting their behaviour in the various biological systems of concern, and consequently are of great importance in specifying when particular studies and tests are required, and in the design of such studies.
|
|
(iv)
|
The extent to which active substances comply with relevant FAO specifications, must be stated. Divergences from FAO specifications must be described in detail, and justified.
|
|
(v)
|
In certain specified instances, tests must be conducted using purified active substance of stated specification. In such cases the method(s) of purification employed and their effectiveness must be reported. The purity of such test material, which must be as high as can be achieved using the best available technology, must be reported. A reasoned justification must be provided in cases where the degree of purity achieved is less than 980 g/kg. The justification provided, must demonstrate that all technically feasible and reasonable possibilities for the production of pure active substance have been exhausted.
|
|
3.1
|
Melting point and boiling point
|
|
3.1.1
|
The melting point or where appropriate the freezing or solidification point of purified active substance must be determined and reported according to EEC Method A 1. Measurements should be taken at temperatures up to 360°C.
|
|
3.1.2
|
Where appropriate, the boiling point of purified active substances must be determined and reported according to EEC Method A 2. Measurements should be taken at temperatures up to 360°C.
|
|
3.1.3
|
Where melting point and/or boiling point cannot be determined because of decomposition or sublimation, the temperature at which decomposition or sublimation occurs, must be reported.
|
|
3.2
|
Relative density
|
|
|
In the case of active substances which are liquids or solids, the relative density of the active substance as manufactured and that of purified active substance must be determined and reported according to EEC Method A 3.
|
|
3.3
|
Vapour pressure (in Pa) at 20°C, volatility (e.g. Henry's law constant)
|
|
3.3.1
|
The vapour pressure of purified active substance, determined in accordance with EEC Method A 4, must be reported. Where vapour pressure is less than 10-5 Pa, the vapour pressure at 20°C may be estimated, using a vapour pressure curve.
|
|
3.3.2
|
In the case of active substances which are solids or liquids, volatility (Henry's law constant) of purified active substance must be calculated from its water solubility and vapour pressure and be reported (in Pa x m3 x mol-1).
|
|
3.4
|
Appearance (physical state, colour and odour; if known)
|
|
3.4.1
|
A description of both the colour, if any, and the physical state of both the active substance as manufactured and of purified active substance, must be provided.
|
|
3.4.2
|
A description of any odour associated with the active substance as manufactured and of purified active substance, noted when handling the materials in laboratories or production plants, must be reported.
|
|
3.5
|
Spectra (UV/VIS, IR, NMR, MS), molecular extinction at relevant wavelengths
|
|
3.5.1
|
The following spectra, including a table of signal characteristics needed for interpretation, must be determined using purified active substance, and be reported ultraviolet/visible (UV/VIS), infrared (IR), nuclear magnetic resonance (NMR), and mass spectra (MS). Molecular extinction at relevant wavelengths, must also be determined and reported. The wavelengths at which UV/VIS molecular extinction occurs are determined and reported must include, where appropriate, a wavelength at the highest absorption value above 290 nm. In the case of active substances which are resolved optical isomers, their optical purity must be measured and reported.
|
|
3.5.2
|
The UV/VIS absorption spectra, IR, NMR and MS spectra, where necessary for the identification of impurities considered to be of toxicological or environmental significance, and molecular extinction at relevant wavelengths, must be determined and reported.
|
|
3.6
|
Solubility in water including effect of pH (4 to 10) on solubility
|
|
|
The water solubility of purified active substances which are not volatile, at 20°C and atmospheric pressure, must be determined in accordance with EEC Method A 6, and be reported. These water solubility determinations must be made in the neutral range (i.e. in purified water in equilibrium with atmospheric carbon dioxide). Where the active substance is capable of forming ions, determinations must also be made in the acidic range (pH 4 to 6) and in the alkaline range (pH 8 to 10), and be reported. Where the stability of the active substance in aqueous media is such that water solubility cannot be determined, a justification based on test data must be provided.
|
|
3.7
|
Solubility in organic solvents
|
|
|
The solubility of active substances, as manufactured, in the following organic solvents at 20°C must be determined and reported — if less than 250 g/kg, the temperature applied must be reported:
|
|
|
Aliphatic hydrocarbon
|
— preferably n-heptane
|
|
|
Aromatic hydrocarbon
|
— preferably xylene
|
|
|
Halogenated hydrocarbon
|
— preferably 1,2-dichloroethane
|
|
|
Alcohol
|
— preferably methanol or isopropyl alcohol
|
|
|
Ketone
|
— preferably acetone
|
|
|
Ester
|
— preferably ethyl acetate
|
|
|
If for a particular active substance, one or more of these solvents is unsuitable (e.g. reacts with test material), alternative solvents can be used instead. In such cases, choices made must be justified in terms of their structure and polarity.
|
|
| | |
3.8
|
Partition coefficient n-octanol/water including effect of pH (4 to 10)
|
|
|
The n-octanol/water partition coefficient of purified active substance at 20°C, must be determined and reported according to EEC Method A 8. The effect of pH (4 to 10) must be investigated when the substance is acidic or basic as defined by its pKa value (2 for bases).
|
|
3.9
|
Stability in water, hydrolysis rate, photochemical degradation, quantum yield and identity of breakdown product(s), dissociation constant including effect of pH (4 to 9)
|
|
3.9.1
|
The hydrolysis rate of purified active substances (usually radiolabelled active substance,>95% purity), at 20°C, for each of the pH values 4, 7 and 9, under sterile conditions, in the absence of light, must be determined in accordance with EEC Method C 7, and be reported. For substances with a low rate of hydrolysis, the rate can be determined at 50°C, or another appropriate temperature. If degradation is observed at 50°C, degradation rate at another temperature must be determined, and an Arrhenius plot must be constructed to permit an estimate to be made of hydrolysis at 20°C. The identity of hydrolysis products formed and the rate constant observed, must be reported. The estimated DT50 value must also be reported.
|
|
3.9.2
|
For compounds with a molar decadic absorption coefficient (ε) > 10 (1 x mol-1 x cm-1) at a wavelength λ >290 nm, direct phototransformation in purified water (e.g. distilled water) at 20 to 25°C, of purified active substance, usually radio labelled, using artificial light (intensities and spectral distribution relevant to northern European), under sterile conditions, if necessary using a solubilizer, must be determined and reported. Sensitizers such as acetone must not be used as a co-solvent or solubilizer. The light source must simulate sunlight and be equipped with filters to exclude radiation at wavelengths λ > 290 nm. The identity of breakdown products formed, which at any time during the study are present in quantities> 10% of the active substance added, must be reported. In addition, a mass balance to account for at least 90% of the radioactivity, as well as photochemical halflife must be reported.
|
|
3.9.3
|
Where necessary to investigate direct phototransformation, the quantum yield of direct photodegradation in water must be determined and reported, together with calculations to estimate theoretical lifetime of the active substance in the top layer of aqueous systems and the real lifetime of the substance. The method to be used is that described in the Annex to the FAO Revised Guidelines on Environmental Criteria for the Registration of Pesticides.
|
|
3.9.4
|
Where dissociation in water occurs, the dissociation constant(s) (pKa) of purified active substances, at 20°C, must be determined and reported in accordance with OECD Test Guideline 112. The identity of the dissociated species formed, based on theoretical considerations, must be reported. If the active substance is a salt, the pKa value of the active principle must be given.
|
|
3.10
|
Stability in air, photochemical degradation, identity of breakdown product(s)
|
|
|
An estimation of the photochemical oxidative degradation (indirect phototransformation) of the active substance(s), must be provided.
|
|
3.11
|
Flammability including auto-flammability
|
|
3.11.1
|
The flammability of active substances as manufactured, which are solids, gasses, or are substances which in contact with water or damp air evolve highly flammable gasses, must be determined in accordance with EEC Method A 10, A 11, or A 12, as appropriate, and be reported.
|
|
3.11.2
|
The auto-flammability of active substances as manufactured, determined in accordance with EEC Method A 15, or A 16, as appropriate, must be reported.
|
|
3.12
|
Flash point
|
|
|
The flash point of active substances as manufactured, which have a melting point below 40°C, must be determined in accordance with EEC Method A 9 using a closed cup method, and be reported.
|
|
3.13
|
Explosive properties
|
|
|
The explosive properties of active substances as manufactured, determined in accordance with EEC Method A 14, must be reported, for substances which on theoretical grounds are potentially explosive.
|
|
3.14
|
Oxidizing properties
|
|
|
The oxidizing properties of active substances as manufactured, determined in accordance with EEC Method A 17, must be reported, except where examination of its structural formula, establishes beyond reasonable doubt that the active substance is incapable of reacting exothermically with a combustible material. In such cases, it is sufficient to provide that information as justification for not determining the oxidizing properties of the substance.
|
|
4
|
Further information on the active substance(s)
|
|
|
The intended function of each active substance must be specified from among the following:
|
|
| | |
acaricide
|
nematicide
|
|
algicide
|
semio chemical
|
|
bactericide
|
plant growth regulator
|
|
defoliant
|
repellant (mammalian, avian,
|
|
fungicide
|
earthworm or insect)
|
|
herbicide (selective)
|
rodenticide
|
|
herbicide (non-selective)
|
talpicide
|
|
insecticide
|
viricide
|
|
molluscicide
|
other (must be specified)
|
|
| | |
5
|
Identity of the plant protection product
|
|
(i)
|
The information provided, together with that provided for active substances concerned, must be sufficient to identify with precision, the plant protection product, to define it in terms of its specification and to characterize it as to its nature. The information and data referred to, unless otherwise specified, are required for all plant protection products.
|
|
(ii)
|
The extent to which plant protection products for which authorization is sought, comply with relevant FAO specifications, must be stated. Divergences from FAO specifications must be described in detail, and justified.
|
|
5.1
|
Manufacturer of the preparation and the active substance(s) (names and addresses, etc. including location of plants)
|
|
5.1.1
|
The name and address of the manufacturer of the preparation must be provided as must the name and address of each manufacturing plant in which the preparation is manufactured. A contact point (preferably a central contact point, to include name, telephone and telefax numbers) must be provided for each.
|
|
5.2
|
Trade name or proposed trade name, and manufacturer's development code number of the preparation if appropriate
|
|
|
All former and current trade names and proposed trade names and development code numbers of the preparation as well as current names and numbers must be provided. Where trade names and code numbers referred to, relate to similar but different preparations (possibly obsolete), full details of the differences, must be provided.
|
|
5.3
|
Detailed quantitative and qualitative information on the composition of the preparation (active substance(s), impurities, adjuvants, inert components, etc.)
|
|
5.3.1
|
The content (minimum, mean and maximum in g/kg for solids and in g/kg and g/l at 20°C for liquids) in the preparation, must be provided:
|
|
|
— for active substances, in terms both of technical active substance and pure active substance; and
|
|
|
— for each formulant, other than active substances.
|
|
|
In the case of active substances, the salt, ester, anion, or cation concerned, where appropriate, must be specified.
|
|
5.3.2
|
Formulants must where possible, be identified both by their chemical name as given in Annex I to the Directive of 1967, or if not included in that Directive, in accordance with both IUPAC and CA nomenclature. Their ISO common name or proposed common name, where relevant, must be provided. Their structure or structural formula must be provided. For each component of a formulant, the relevant EEC (EINECS or ELINCS) number and CAS number, where they exist, must be provided. Where the information provided does not fully identify a formulant or a component of a formulant, an appropriate specification must be provided. The trade name of both formulants and their components, where they exist, must also be provided.
|
|
5.3.3
|
For formulants, other than active substance, their function must be given:
|
|
| | |
adhesive (sticker)
|
dye
|
repellent
|
|
antifoaming agent
|
emetic
|
safener
|
|
antifreeze
|
emulsifier
|
solvent
|
|
binder
|
fertiliser
|
stabiliser
|
|
buffer
|
odourant
|
synergist
|
|
carrier (solid)
|
perfume
|
thickener
|
|
deodorant
|
preservative
|
wetting agent
|
|
dispersing agent
|
propellant
|
miscellaneous (specify)
|
|
| | |
5.4
|
Physical state and nature of the preparation (emulsifiable concentrate, wettable powder, solution etc.)
|
|
|
The type of preparation must be designated according to the coding system contained in Annex 2 to Part 2 of the Fourth Schedule. Where a particular preparation is not defined in that system, a full description of the physical nature and state of the preparation must be provided, together with a proposal for a suitable description of the type of preparation and a proposal for its definition.
|
|
5.5
|
Function (herbicide, insecticide, etc.) of the plant protection product
|
|
|
The intended function, or functions, of the preparation must be specified from among the following:
|
|
| | |
acaricide
|
nematicide
|
|
algicide
|
semio chemical
|
|
bactericide
|
plant growth regulator
|
|
defoliant
|
repellant (mammalian, avian,
|
|
fungicide
|
earthworm or insect)
|
|
herbicide (selective)
|
rodenticide
|
|
herbicide (non-selective)
|
talpicide
|
|
insecticide
|
viricide
|
|
molluscicide
|
other (must be specified)
|
|
| | |
6.
|
Physical and chemical properties of the plant protection product
|
|
6.1
|
Appearance (physical state, colour and odour; if known)
|
|
|
A description of both the colour, if any, and the physical state of the preparation, must be provided.
|
|
6.2
|
Explosivity and oxidizing properties
|
|
6.2.1
|
The explosive properties of preparations, determined in accordance with EEC Method A 14, must be reported, for substances which on theoretical grounds are potentially explosive. Where available thermodynamic information serves to establish beyond reasonable doubt that the preparation is incapable of exothermic reaction, it is sufficient to provide that information as a justification for not determining the explosive properties of the preparation.
|
|
6.2.2
|
The oxidizing properties of preparations, determined in the case of those which are solids, in accordance with EEC Method A 17, must be reported. In other cases, the method used must be justified. The oxidizing properties of preparations need not be determined where it can be shown, without reasonable doubt, on the basis of thermodynamic information, that the preparation is incapable of reacting exothermically with a combustible material. In such cases, it is sufficient to provide that information as justification for not determining the oxidizing properties of the substance.
|
|
6.3
|
Flash point and other indications of flammability or spontaneous ignition
|
|
|
The flash point of liquids which contain flammable organic solvents, must be determined in accordance with EEC Method A 9, and be reported. The flammability of solid preparations and gasses must be determined in accordance with EEC Method A 10, A 11, A 12, as appropriate, and be reported. In addition the auto-flammability of preparations must be determined in accordance with EEC Method A 15 or A16, as appropriate, and be reported.
|
|
6.4
|
Acidity / alkalinity and if necessary pH value
|
|
6.4.1
|
In the case of preparations which are acidic (pH 4) or alkaline (pH 10), their acidity or alkalinity and the pH value must be determined in accordance with CIPAC Method MT 31 and MT 75, as appropriate, and be reported.
|
|
6.4.2
|
Where relevant (if to be applied as an aqueous dilution) the pH of a 1% aqueous dilution, emulsion or dispersion of the preparation, must be determined in accordance with CIPAC Method MT 75, and be reported.
|
|
6.5
|
Viscosity and surface tension
|
|
6.5.1
|
In the case of liquid preparations for ultra low volume application (ULV), the kinematic viscosity of the preparation must be determined in accordance with OECD Test Guideline 114, and be reported.
|
|
6.5.2
|
For non newtonian liquids the viscosity must be determined and reported together with the test conditions.
|
|
6.5.3
|
In the case of liquid preparations, the surface tension of the preparation must be determined in accordance with EEC Method A 5, and be reported.
|
|
6.6
|
Relative density
|
|
|
In the case of preparations which are liquids or solids, the relative density of the preparation must be determined in accordance with EEC Method A 3, and be reported.
|
|
6.7
|
Reactivity towards container materials
|
|
|
The resistance of the proposed packaging material to its contents must be determined and reported.
|
|
7.
|
Information on the proposed experimental use
|
|
7.1
|
Field of use e.g. field, glasshouse, food or feed storage, home garden
|
|
|
The field of proposed use must be specified from among the following:
|
|
|
Crop Protection (Agriculture and Horticulture)
|
|
|
—Field crops, pre-harvest
|
|
|
—Field crops, post-harvest
|
|
|
—Protected crops, pre-harvest
|
|
|
—Protected crops, post-harvest
|
|
|
Forestry —Nursery use
|
|
|
—Crop protection
|
|
|
—Timber protection
|
|
|
Home Garden Use
|
|
|
Household Use
|
|
|
Food and feed storage practice
|
|
|
Non-selective Weed Control —Agriculture and horticulture
|
|
|
—Home garden
|
|
|
—Industrial
|
|
|
—Amenity
|
|
|
Other Amenity Use —Plant protection
|
|
|
—Aquatic weed control
|
|
|
—Algae control
|
|
|
Other (specify)
|
|
7.2
|
Effects on harmful organisms, e.g. contact, inhalation or stomach poison, fungitoxic or fungistatic, etc., systemic or not in plants
|
|
3.2.1
|
The nature of the effects on harmful organisms must be stated:
|
|
|
contact action
|
|
|
stomach action
|
|
|
inhalation action
|
|
|
fungitoxic action
|
|
|
fungistatic action
|
|
|
desiccant
|
|
|
reproduction inhibitor
|
|
|
other (must be specified)
|
|
3.2.2
|
Where relevant it must be stated, for each active substance in the preparation, whether or not the active substance is translocated in plants and where relevant whether such translocation is apoplastic, symplastic, or both.
|
|
7.3
|
Details of intended use e.g. types of harmful organisms controlled and/or plants or plant products to be protected
|
|
7.3.1
|
Details of the intended use in terms of crops, groups of crops, plants, or plant products protected, must be provided.
|
|
7.3.2
|
Details of the harmful organisms to be controlled, and where relevant, details of harmful organisms against which protection is afforded, must be provided.
|
|
7.3.3
|
Where relevant, details of other effects to be achieved e.g. sprout suppression, retardation of ripening, reduction in stem length, enhanced fertilization etc., must be provided.
|
|
7.4
|
Proposed application rate
|
|
|
For each method of application and each use, the rate of application per unit treated, in terms of g or kg of both product and active substance, must be provided. Application rates should be expressed in g or kg/hectare for field crops, in g or kg/100 m2 for glasshouse crops and home garden uses, in g or kg/tonne for treated seed, and in g or kg/unit or unit volume in other cases, as appropriate.
|
|
7.5
|
Concentration of active substance in material used (e.g. in the diluted spray, baits or treated seed)
|
|
|
The content of active substance in g/l of diluted spray material, in g/kg or mg/kg of bait or in g/tonne of treated seed, as appropriate, must be reported.
|
|
7.6
|
Proposed method of application
|
|
|
The method of application proposed must be described fully, indicating the type of equipment to be used, if any, as well as the type and volume of diluent to be used per unit area or volume.
|
|
7.7
|
Number and timing of applications proposed and duration of protection
|
|
7.7.1
|
The maximum number of applications to be used and their timing, must be reported. Where relevant the growth stages of the crop or plants to be protected, and where relevant the development stage of the harmful organism concerned, or their condition, must be indicated. Where possible the interval between applications, in days, must be stated.
|
|
7.7.2
|
Where relevant, the anticipated duration of protection afforded both by each application and by the maximum number of applications to be used, must be indicated.
|
|
7.8
|
Necessary waiting periods or other precautions to avoid Phytotoxic effects on succeeding crops
|
|
7.8.1
|
Where relevant and known, minimum waiting periods between last application and sowing or planting of succeeding crops, which are necessary to avoid phytotoxic effects on succeeding crops, must be stated.
|
|
7.8.2
|
Known limitations on choice of succeeding crops, if any, must be stated.
|
|
7.9
|
Re-entry periods, necessary waiting periods or other precautions to protect man, livestock and the environment
|
|
|
Where relevant, crop destruction, pre-harvest intervals, re-entry periods, withholding periods, or other precautions necessary to minimize the presence of residues in or on crops, plants and plant products, or in treated areas or spaces, with a view to protecting man, livestock or the environment, must be specified:
|
|
|
— crop destruction;
|
|
|
— pre-harvest interval (in days) for each relevant crop;
|
|
|
— re-entry period (in days) for livestock, to areas to be grazed;
|
|
|
— re-entry period (in hours or days) for man to crops, buildings or spaces treated;
|
|
|
— withholding period (in days) for animal feedingstuffs;
|
|
|
— waiting period (in days), between application and handling treated produce;
|
|
|
— waiting period (in days), between last application and sowing or planting succeeding crops; or
|
|
|
— other precautions, which must be described.
|
|
7.10
|
Proposed instructions for use
|
|
|
The proposed instructions for use of the preparation, to be followed in the application of the product, must be provided.
|
|
8.
|
Analytical methods
|
|
|
Where use is on a crop or plant product intended for use as food or feed, if residues may occur (moderate or high risk) and the crop or plant product is not to be destroyed, relevant, analytical methods including recovery rates and the limits of determination for residues in treated plants, plant products, foodstuffs and feeding-stuffs, must be provided.
|
|
9
|
Efficacy data
|
|
9.1
|
The data supplied must be sufficient to permit an evaluation to be made of the potential role of the plant protection product, for the uses proposed. In particular it must be possible to establish that on the basis of the information available, it is possible that the product may fulfil its intended purpose, in terms of control of harmful organisms or other intended effects.
|
|
9.2
|
Reports in summary form of preliminary tests (primary biological screening), including glasshouse and field studies, used to assess the biological activity and dose range finding of the plant protection product and of the active substance(s) it contains, must be submitted. Where this information is not available, a reasoned statement of the rational used to suggest that the plant protection product may fulfil its intended purpose, in terms of control of harmful organisms or other intended effects, must be provided.
|
|
10.
|
Toxicological studies
|
|
10.1
|
Acute toxicity
|
|
(i)
|
The studies, data and information to be provided and evaluated, must be sufficient to permit the hazards for humans, following a single exposure to the plant protection product, to be assessed, and in particular to establish, or indicate:
|
|
|
— the relationship between dose and morbidity and mortality;
|
|
|
— the toxicity of the active substance relative to other substances;
|
|
|
— the time course and characteristics of poisoning with full details of behaviourial changes and possible gross pathological findings at post-mortem; and
|
|
|
— the relative hazard associated with the different routes of exposure.
|
|
(ii)
|
While the emphasis must be on estimating the toxicity ranges involved, the information generated must also permit the active substance(s) and the preparation to be classified. The information generated through acute toxicity testing is of particular value in assessing hazards likely to arise in accident situations.
|
|
(iii)
|
The vehicle chosen for administration of the test material must be selected such that it is not itself toxic and such that the test substance is bio-available. Where feasible, aqueous solutions are preferred. Other options include solution/suspension in 0.5% carboxymethyl cellulose or solution/suspension in non-polar non-toxic solvents. Where a novel or unusual vehicle is used, the bio-availability of the test substance in the vehicle used must be established and reported —a comparative 7 day test using radiolabelled material with different vehicles.
|
|
10.1.1
|
Oral
|
|
10.1.1.1
|
The acute oral toxicity to the rat of each active substance in the preparation, determined in accordance with the EEC Method B 1 (Fixed Dose Method), must be reported.
|
|
10.1.1.2
|
The acute oral toxicity to the rat of the preparation, determined in accordance with the EEC Method B 1 (Fixed Dose Method), must be reported, except where:
|
|
|
— for plant protection products containing one active substance, Article 3.2 of the Directive of 1978 can in principle be invoked; or
|
|
|
— for plant protection products containing more than one active substance, Article 3.3 of the Directive of 1978 can in principle be invoked; and
|
|
|
relevant information and documentation is submitted, or is available to the competent authority, which shows that there are valid grounds for assuming that the classification resulting from the calculation would not vary substantially from that obtainable by biological testing i.e. the composition of the preparation is similar to that of a preparation for which a biological test is available, and the toxicity of the tested preparation is similar to its predicted toxicity, or the composition of the preparation is similar to that of two or more preparations for which biological tests are available, and the toxicity of the tested preparations varies in a consistent manner from their predicted toxicities.
|
|
10.1.2
|
Percutaneous
|
|
10.1.2.1
|
Since knowledge of percutaneous toxicity is necessary for the evaluation of operator hazard and the specification of suitable protective clothing, the acute percutaneous toxicity to rats of each active substance in the preparation, determined in accordance with EEC Method B 3, must be reported. Both local and systemic effects must be investigated.
|
|
10.1.2.2
|
The acute percutaneous toxicity to rats of the preparation, determined in accordance with EEC Method B 3, must be reported. Both local and systemic effects must be investigated. However, testing of the preparation should not be carried out, where :
|
|
|
— for plant protection products containing one active substance, Article 3.2 of the Directive of 1978 can in principle be invoked; or
|
|
|
— for plant protection products containing more than one active substance, Article 3.3 of the Directive of 1978 can in principle be invoked; and
|
|
|
relevant information and documentation is submitted, or is available to the competent authority, which shows that there are valid grounds for assuming that the classification resulting from the calculation would not vary substantially from that obtainable by biological testing i.e. the composition of the preparation is similar to that of a preparation for which a biological test is available, and the toxicity of the tested preparation is similar to its predicted toxicity, or the composition of the preparation is similar to that of two or more preparations for which biological tests are available, and the toxicity of the tested preparations varies in a consistent manner from their predicted toxicities.
|
|
10.1.3
|
Inhalation
|
|
10.1.3.1
|
The inhalation hazard of a plant protection product is dependant not only on its toxic properties, physical state, manner of use and where relevant particle size, but also on the vapour pressure and solubility of its components. Accordingly, the inhalation toxicity to rats of each active substance in the preparation, determined in accordance with EEC Method B 2, must be reported where the preparation is:
|
|
|
— a gas or liquified gas;
|
|
|
— to be used as a fumigant;
|
|
|
— to be used with fogging equipment;
|
|
|
— to be included in a smoke generating, aerosol or vapour releasing preparation;
|
|
|
— to be included in preparations which are powders containing a significant proportion of particles of diameter1% on a weight basis); or
|
|
|
— to be included in preparations to be applied in a manner which produces a spray, mist or aerosol containing a significant proportion of particles of diameter1% on a weight basis).
|
|
10.1.3.2
|
In addition, in the case of active substances having a vapour pressure> 1 x 10-2 Pa, and which are to be included in preparations to be used in enclosed spaces such as warehouses or glasshouses, the inhalation toxicity to rats of each such active substance, determined in accordance with EEC Method B 2, must be reported.
|
|
10.1.3.3
|
The inhalation toxicity to rats of the preparation, determined in accordance with EEC Method B 2, must be reported where the preparation is:
|
|
|
— a gas or liquified gas;
|
|
|
— a smoke generating formulation or a fumigant;
|
|
|
— a vapour releasing preparation;
|
|
|
— to be used with fogging equipment;
|
|
|
— contains active substance(s) having a vapour pressure>1 x 10-2 Pa and to be included in preparations to be used in enclosed spaces such as warehouses or glasshouses;
|
|
|
— to be included in a smoke generating, aerosol or vapour releasing preparation;
|
|
|
— to be included in preparations which are powders containing a significant proportion of particles of diameter 1% on a weight basis); or
|
|
|
— to be included in preparations to be applied in a manner which produces a spray, mist or aerosol containing a significant proportion of particles of diameter1% on a weight basis).
|
|
10.1.3.4
|
However, testing of the preparation should not be carried out, where :
|
|
|
— for plant protection products containing one active substance, Article 3.2 of the Directive of 1978 can in principle be invoked; or
|
|
|
— for plant protection products containing more than one active substance, Article 3.3 of the Directive of 1978 can in principle be invoked; and
|
|
|
relevant information and documentation is submitted, or is available to the competent authority, which shows that there are valid grounds for assuming that the classification resulting from the calculation would not vary substantially from that obtainable by biological testing i.e. the composition of the preparation is similar to that of a preparation for which a biological test is available, and the toxicity of the tested preparation is similar to its predicted toxicity, or the composition of the preparation is similar to that of two or more preparations for which biological tests are available, and the toxicity of the tested preparations varies in a consistent manner from their predicted toxicities.
|
|
10.1.4
|
Skin and where appropriate eye irritation
|
|
10.1.4.1
|
The possible effects of accidental contamination of skin and eyes, must be investigated, as contact may occur in handling preparations containing the active substance, in either concentrated or dilute form, or in cleaning application equipment.
|
|
10.1.4.2
|
The skin irritancy of each active substance, and of the preparation, determined using a single application to intact skin of rabbits, in accordance with EEC Method B 4, must be reported, except where:
|
|
|
— positive results can be predicted on the basis of the acidity or alkalinity of aqueous solutions of the active substance (pH S2 or> 11.5), or on the basis of the acidity or alkalinity of the preparation (pH 2> or11.5), as appropriate;
|
|
|
— no irritation is seen at the limit test dose level in acute percutaneous studies; or
|
|
|
— where the active substance, or preparation, as appropriate, is highly toxic in an acute percutaneous study.
|
|
10.1.4.3
|
Eye irritation tests must not be conducted for active substances or preparations known, or found, to be corrosive. Similarly eye irritation tests must not be conducted for substances or preparation known or found to be skin irritants, unless there is particular reason to believe otherwise. For other active substances contained in the preparation, as well as for the preparation, eye irritancy, determined in accordance with EEC Method B 5, using healthy adult albino rabbits, must be determined and reported, except where it is known, on the basis of other available information that the active substance or preparation, as appropriate, is likely to produce severe effects on the eyes.
|
|
10.1.5
|
Skin sensitization
|
|
10.1.5.1
|
Allergic sensitization, following exposure, occurs in a significant proportion of the human population. The potential of each active substance in the preparation, to provoke skin sensitization reactions, must be assessed in accordance with the EEC Method B 6, using the Guinea-pig Maximization Test (GPMT), and be reported.
|
|
10.1.5.2
|
The potential of preparations which contain an active substance found to provoke skin sensitization reactions, or which contain other components known to lead to such reactions, must be assessed in accordance with the EEC Method B 6, and be reported.
|
|
10.2
|
Short-term toxicity
|
|
(i)
|
Short-term toxicity studies to be submitted and evaluated, must be designed to provide information as to the amount of the active substance that can be tolerated without toxic effects under the conditions of the study. Such studies provide useful data on the risks for those handling and using preparations containing the active substance. In particular, short-term studies provide an essential insight into possible cumulative actions of the active substance, and the risks to workers who may be intensively exposed. In addition short-term studies provide information useful in the design of chronic toxicity studies.
|
|
(ii)
|
The studies, data and information to be provided and evaluated, must be sufficient to permit the hazards for humans, following repeated exposure to the active substance, to be assessed, and in particular to further establish, or indicate:
|
|
|
— the relationship between dose and adverse effects;
|
|
|
— toxicity of the active substance relative to other substances;
|
|
|
— target organs, where relevant;
|
|
|
— the time course and characteristics of poisoning with full details of behaviourial changes and possible pathological findings at post-mortem;
|
|
|
— specific toxic effects and pathological changes produced;
|
|
|
— where relevant, the persistence and reversibility of certain toxic effects observed, following discontinuation of dosing;
|
|
|
— where possible, the mode of toxic action; and
|
|
|
— the relative hazard associated with the different routes of exposure.
|
|
(iii)
|
The studies specified are always required in support of applications for authorization for trials purposes where the nature of the proposed use is such that a there is a moderate or high risk of residues occurring in food or feed commodities, and where it is not proposed that produce be destroyed.
|
|
10.2.1
|
Repeated dose oral toxicity (28-day study)
|
|
10.2.1.1
|
Generally, range finding short-term (28 day) studies are an optional requirement. Where conducted they must be reported, since the results can be of particular value in the identification of adaptive responses which can be masked in testing over longer periods. Testing is required where the structure of the active substance (structure activity relationship), or the results of acute toxicity testing, suggest neurotoxic potential.
|
|
10.2.1.2
|
The repeated dose oral toxicity to rats of each active substance in the preparation, following exposure for 28 days, where relevant, must be determined, and reported. The appropriate test guideline is EEC Method B 7.
|
|
10.2.2
|
Oral 90-day study —two species
|
|
10.2.2.1
|
The short-term oral toxicity (90 day) of each active substance in the preparation, to both rat and dog (of defined breed), determined in accordance with the methods specified in Commission Directive 87/302/EEC34 (Sub-chronic oral toxicity test: 90-day repeated oral dose using rodent species, and Sub-chronic oral toxicity test: 90-day repeated oral dose using non-rodent species), must be reported. Where there is evidence of species differences, and where such data are likely to be of value in extrapolating results obtained to man, a 12 month short-term toxicity study in dogs should be conducted and reported.
|
|
10.2.2.2
|
The number of test animals used in the rat study must, where appropriate, be sufficient to facilitate the determination of the persistence and reversibility of observed effects, following the discontinuation of dosing, as well as to permit observation of delayed effects, if any. Except where it is self evident that effects are persistent and are not reversible, the persistence and reversibility of observed effects, as well as the delayed occurrence of toxic effects, must be reported, unless the effects only occur at doses higher than the potential levels of human exposure, taking account of the appropriate and required margins of safety.
|
|
|
34 O. J. No. L 133/1 30/5/1988.
|
|
10.3
|
Genotoxicity —test strategy to assess gene mutations, chromosomal aberrations and DNA perturbations
|
|
(i)
|
Genotoxicity studies are of value in;
|
|
|
— the prediction of heritable defects;
|
|
|
— the early identification of genotoxic carcinogens; and in
|
|
|
— the elucidation of the mechanism of action of some carcinogens.
|
|
(ii)
|
A sequential, or tiered, approach to testing is specified (Figure 4). To avoid responses that are artifacts of the test system, excessively toxic doses must not be used in either in vitro or in vivo assays for genotoxicity.
|
|
(iii)
|
The studies specified are always required in support of applications for authorization for trials purposes where the nature of the proposed use is such that a there is a moderate or high risk of residues occurring in food or feed commodities, and where it is not proposed that produce be destroyed. Testing can be suspended where on completion of Tier 1 or Tier 2 testing, as appropriate, the results obtained are negative.
|
|
10.3.1
|
Tier 1 In Vitro Testing
|
|
10.3.1.1
|
All in vitro screening tests must be duplicated in separate experiments to confirm the results obtained. Where appropriate, different concentrations of active substance should be investigated in such "repeat" studies. Spurious positive results which are "stress-related" rather than indicative of intrinsic mutagenicity, can result from low pH and high osmolarity in assay systems.
|
|
10.3.1.2
|
Screening studies for gene mutations (base-pair and frame shift) in Salmonella typhimurium, strains TA98, TA100, TA102, TA1535, and TA1537, Escherichia coli WP2 andEscherichia coli WP2 uvrA, conducted in accordance with EEC Methods B 13 and B 14, as appropriate, to include testing in the presence and absence of exogenous metabolic activation, (Figure 4), must be reported.
|
|
10.3.1.3
|
In vitro tests for clastogenicity, to detect structural chromosomal aberrations, using human lymphocytes, conducted in accordance with the EEC Method B 10, must be reported (Figure 4).
|
|
10.3.1.4
|
Since the bacterial gene mutation and the mammalian cytogenetic assays, while efficient, do not detect all compounds with mutagenic potential, further testing for gene mutation in mammalian cells must be conducted, and reported (Figure 4). Tests must be conducted in accordance with the method specified in Commission Directive 87/302/EEC (In vitro mammalian cell gene mutation test) using L5178Y mouse lymphoma cells (TK locus), or other cell lines shown to be as sensitive.
|
|
| |
Figure 4
|
| |
GENOTOXICITY TESTING
|
| |
Criteria to determine when tests are required
|
| |
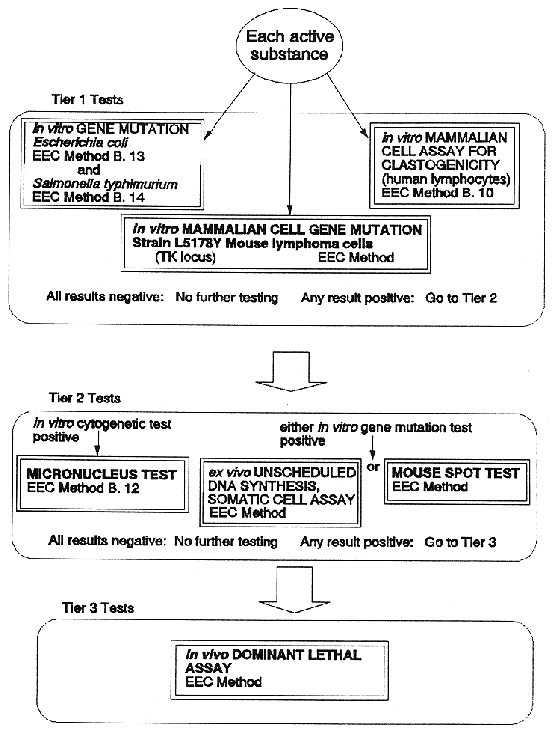
|
| | |
10.3.2
|
Tier 2 In Vivo Testing, Somatic Cells
|
|
10.3.2.1
|
Where the results of any of the in vivo cytogenetic test is positive, an in vitro test using somatic cells, in accordance with EEC Method B 12, is required (Figure 4). The route of administration, usually oral, must be chosen, to ensure significant absorption in test animals.
|
|
10.3.2.2
|
Where the results of either of the in vitro gene mutation tests are positive, a further test to investigate unscheduled DNA synthesis in rat liver ex vivo, in accordance with the method specified in Commission Directive 87/302/EEC (DNA damage and repair - unscheduled DNA synthesis —mammalian cells in vitro) or a mouse spot test conducted in accordance with the method specified in Commission Directive 87/302/EEC (Mouse spot test), must be reported.
|
|
10.3.3
|
Tier 3 In Vivo Testing, Germ Cells
|
|
|
Where an active substance is found to be genotoxic in vivo in somatic cells, genotoxic potential in germ cells must also be investigated (Figure 4), since not all somatic cell mutagens are germ cell mutagens. Except where use of an alternative method is justified, a dominant lethal assay, conducted in accordance with the method specified in Commission Directive 87/302/EEC (Rodent dominant lethal test), must be reported. The route of administration, usually oral intubation, must be chosen, to ensure significant absorption in test animals.
|
|
11
|
Residues in or on treated products, food or feed
|
|
(i)
|
In principle, the information provided, must be sufficient to permit an evaluation to be made as to the level, nature, and identity of residues, from time of application to harvest, slaughter, or collection, or following storage as appropriate, and as to the decline of such residues, where relevant.
|
|
(ii)
|
In principle, the information provided, must be sufficient to permit an evaluation to be made as to potential exposure, through diet of consumers, to residual traces of active substance and toxicologically significant metabolites, degradation and reaction products. Taken together with relevant and available toxicological information, it must, in principle, be sufficient to permit an evaluation to be made as to:
|
|
|
— potential exposure of workers, through contact with dislodgeable residues; and
|
|
|
— the need for crops to be destroyed to preclude consumer exposure to residues; or
|
|
|
— the acceptability of proposed pre-harvest, waiting, withholding and re-entry intervals or periods, or other precautions to protect humans and animals.
|
|
(iii)
|
In practice, where in accordance with the rules relating to Comparability, Extrapolation, Group Tolerances and Data Requirements, as contained in Part 3 of the Fifth Schedule, the proposed use is such that there is a low residue risk (see point 5.4.3 of Part 3 of the Fifth Schedule), it is not necessary that data relating to residues at harvest be generated. However, any available data must nevertheless be provided. Where in accordance with those rules, there is a high or medium residue risk (see points 5.4.1 and 5.4.2 of Part 3 of the Fifth Schedule), data relating to residues at harvest must be provided, unless it is proposed that crops be destroyed prior to harvest, or treated plant products be destroyed to prevent consumption, as appropriate.
|
|
11.1
|
Nature of the residue
|
|
11.1.1
|
In the case of plant protection products cleared in accordance with the Regulations of 1994, authorized in accordance with Regulations 13, 15, or 18, for use on crops used for food or feed, evaluated by the WHO/FAO Joint Meeting on Pesticide Residues (JMPR), or evaluated as part of the first, second or subsequent priority groups for the establishment of a Community MRL, a statement must be provided relating to the:
|
|
|
(i) identification of breakdown and reaction products and of metabolites in treated plants or plant products;
|
|
|
(ii) identification of breakdown and reaction products and of metabolites in succeeding crops, where relevant;
|
|
|
(iii) overall material balance for each active substance;
|
|
|
(iv) effects of industrial processing and/or household preparation on the nature and magnitude of residues, where relevant;
|
|
|
(v) results of feeding and metabolism studies in livestock (if residues remain in or on crops or parts of crops used for feed) to permit evaluation of residues in foodstuffs of animal origin; and
|
|
|
(vi) definition of the residue —for each active substance.
|
|
11.1.2
|
In other cases sufficient data must be submitted to identify breakdown and reaction products and metabolites in treated plants or plant products, to establish the effects of industrial processing and/or household preparation on the nature and magnitude of residues, where relevant, and to permit the composition of the residue to be defined.
|
|
12.1
|
Residue levels in treated products, food or feed
|
|
12.1.1
|
Where on the basis of the rules relating to Comparability, Extrapolation, Group Tolerances and Data Requirements, as contained in Part 3 of the Fifth Schedule, the residue levels likely to occur can be estimated on the basis of comparability, the rational and figures used must be provided, together with details of the GAPs used as a basis for the comparison (see Annex 4 of Part 2 of the Fourth Schedule), and the detailed residue data, in summary form, for the crops with which the comparison is made (see Annex 8 of Part 2 of the Fourth Schedule).
|
|
12.1.2
|
In other cases, sufficient residue data from supervised trials in crops, or plant products, for which authorization for trials purposes is sought, must be provided, giving all experimental conditions and details, including residue data concerning the active substance, relevant metabolites and relevant other constituents of the plant protection product, from time of application until harvest, or in the case of post-harvest treatment, breakdown of residues during storage and levels of residues at time of release from storage for marketing.
|
|
| |
PART B
|
| |
Micro-organisms and Viruses
|
| |
(This part does not apply to GMOs where points come under Directive 90/220/EEC)
|
| |
Pro Memoria
|
| |
TENTH SCHEDULE
|
| |
PART 1
|
| |
Regulations 26 (2)
|
| |

|
| |

|
| |

|
| |
PART 2
|
| |
Regulations 26 (7)
|
| |

|
| |

|
| |
PART 3
|
| |
Regulations 26 (7)
|
| |

|
| |

|
| |
ELEVENTH SCHEDULE
|
| |
PART 1
|
| |
Regulations 27 (1)
|
| |

|
| |

|
| |
PART 2
|
| |
Regulations 27 (2)
|
| |

|
| |

|
| |
|
| |
|
| |

|
| |

|
| |
PART 3
|
| |
Regulations 27 (3)
|
| |

|
| |

|
| |

|
| |
THIRTEENTH SCHEDULE
|
| |
Regulations 36 (1)
|
| |
PART 1
|
| | |
Fees for the consideration of applications for the authorization of plant protection products in accordance with Regulations 13, 15 and 18, and for their renewal in accordance with Regulation 19(1)
|
|
|
|
Column (1)
|
Column (2)
|
|
|
£
|
|
Each dossier as specified in Regulation 8 (3) (b)
|
3,500
|
|
Each dossier as specified in Regulation 8 (3) (a)
|
1,250
|
|
Each dossier as specified in Regulation 8 (6)
|
500
|
|
| |
PART 2
|
| | |
Regulations 36 (1)
|
|
Fees for the consideration of applications for modification of authorization for plant protection products in accordance with Regulation 19 (4)
|
|
|
|
|
Column (1)
Category
|
Column (2)
|
|
I
II
|
£
1,250
500
|
|
| | |
"Category I"
|
means a modification in the authorization of a plant protection product involving a major additional use, a major change in the manner of use, or a major formulation change;
|
|
"Category II"
|
means a modification in the authorization of a plant protection product involving a minor additional use, a minor change in the manner of use, or a minor formulation change.
|
|
| |
PART 3
|
| |
Regulations 36 (4)
|
| | |
Annual Fees:
|
£100
|
|
Late Annual Fees:
|
£150
|
|
| |
Given under my Official Seal, this 12th day of May, 1994
|
| |
JOE WALSH,
|
| |
Minister for Agriculture, Food and Forestry
|
| |
EXPLANATORY NOTE
|
| |
These Regulations specify the requirements and conditions for the authorization of plant protection products, which must be complied with in relation to their placing on the market and use, in accordance with Council Directive 91/414/EEC as amended, as well as introducing relevant enforcement and financial provisions.
|
| |
These Regulations also serve to exempt plant protection products which are pesticides regulated in accordance with the European Communities (Classification, Packaging and Labelling of Pesticides) Regulations, 1994 (
S.I. No. 138 of 1994
) from the requirements of those Regulations which require the separate submission of information, data and materials.
|
| |
TWELFTH SCHEDULE
|
| |
Regulation 30 (6)
|
| |
PART 1
|
| | |
Certificate of Result of Analysis.
|
|
Laboratory Reference Number............................................................ ............
|
|
Sample of............................................................ ............................................................ ....................
|
|
received by the designated chemist on............................................................ .............................
|
|
from............................................................ ............................................................ .............................
|
|
Methods of analysis used............................................................ ..................................................
|
|
............................................................ ............................................................ ....................................
|
|
............................................................ ............................................................ ....................................
|
|
This is to certify that the above mentioned sample, which was duty fastened and sealed has been analyzed under the provisions of the European Communities (Authorization, Placing on the Market, Use and Control of Plant Protection Products) Regulations, 1994 (
S.I. No. 139 of 1994
) and the results of the analysis are as follows:—
|
|
............................................................ ............................................................ ....................................
|
|
............................................................ ............................................................ ....................................
|
|
............................................................ ............................................................ ....................................
|
|
............................................................ ............................................................ ....................................
|
|
............................................................ ............................................................ ....................................
|
|
............................................................ ............................................................ ....................................
|
|
............................................................ ............................................................ ....................................
|
|
This certificate is issued under the European Communities (Authorization, Placing on the Market, Use and Control of Plant Protection Products) Regulations, 1994 (
S.I. No. 139 of 1994
).
|
|
Date............................................................ ......
|
Signed............................................................ ...
|
|
|
Designated Chemist.
|
|
| |
PART 2
|
| |
Regulation 34 (2)
|
| |
Certificate of Result of Analysis.
|
| | |
Laboratory Reference Number............................................................
|
|
Sample of............................................................ ............................................................ ........................
|
|
taken at the premises of..........................
|
on............................................................ ..........
|
|
temperature and place of storage............................................................ ..........................................
|
|
Date......................................................
|
Signed............................................................ .......
|
|
|
Authorized Officer
|
|
received by the State Chemist on............................................................ .........................................
|
|
from............................................................ ............................................................ ...............................
|
|
Methods of analysis used............................................................ ....................................................
|
|
............................................................ ............................................................ ......................................
|
|
............................................................ ............................................................ ......................................
|
|
This is to certify that the above mentioned sample, which was duly fastened and sealed, has been analyzed under the provisions of the European Communities (Authorization, Placing on the Market, Use and Control of Plant Protection Products) Regulations, 1994 (
S.I. No. 139 of 1994
) and the results of the analysis are as follows:—
|
|
............................................................ ............................................................ ......................................
|
|
............................................................ ............................................................ ......................................
|
|
............................................................ ............................................................ ......................................
|
|
............................................................ ............................................................ ......................................
|
|
............................................................ ............................................................ ......................................
|
|
............................................................ ............................................................ ......................................
|
|
............................................................ ............................................................ ......................................
|
|
This certificate is issued under the European Communities (Authorization, Placing on the Market, Use and Control of Plant Protection Products) Regulations, 1994 (
S.I. No. 139 of 1994
).
|
|
Date............................................................ .....
|
Signed...........................................................
|
|
|
State Chemist.
|
|