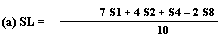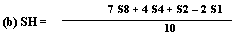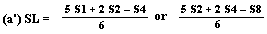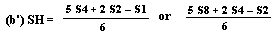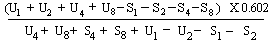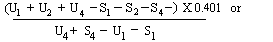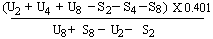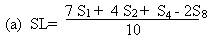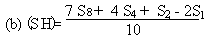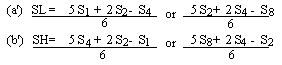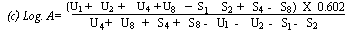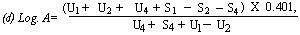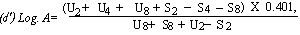S.I. No. 261/1982 - European Communities (Feeding Stuffs) (Methods of Analysis) (Amendment) Regulations, 1982.
I, BRIAN LENIHAN, Minister for Agriculture, in exercise of the powers conferred on me by section 3 of the European Communities Act, 1972 (No. 27 of 1972), for the purpose of giving effect to Commission Directive No. 81/680/EEC of 30 July, 1981,1 and Commission Directive No. 81/715/EEC of 31 July, 1981,2 hereby make the following regulations: | ||||||||||||||||||||||||||||||||||||||||||||
1O.J. L246/32, 29.8.81. | ||||||||||||||||||||||||||||||||||||||||||||
2O.J. L257/38, 10.9.81. 1. (1) These Regulations may be cited as the European Communities (Feeding Stuffs) (Methods of Analysis) (Amendment) Regulations, 1982. | ||||||||||||||||||||||||||||||||||||||||||||
(2) The principal Regulations, Regulation 2 of the Regulations of 1980 and these Regulations may be cited together as the European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1978 to 1982. 2. In these Regulations— | ||||||||||||||||||||||||||||||||||||||||||||
"the Principal Regulations" means the European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1978 ( S.I. No. 250 of 1978 ); | ||||||||||||||||||||||||||||||||||||||||||||
"The Regulations of 1980" means the European Communities (Feeding Stuffs) (Methods of Analysis (Amendment) and Methods of Sampling) Regulations, 1980 ( S.I. No. 14 of 1980 ). 3. The Principal Regulations are hereby amended by— | ||||||||||||||||||||||||||||||||||||||||||||
( a ) the substitution of the following paragraph for paragraph 1 (General Provisions) of Part II: | ||||||||||||||||||||||||||||||||||||||||||||
"1. GENERAL PROVISIONS ON METHODS OF ANALYSIS FOR FEEDING STUFFS. | ||||||||||||||||||||||||||||||||||||||||||||
A. PREPARATION OF SAMPLES FOR ANALYSIS. | ||||||||||||||||||||||||||||||||||||||||||||
1. Purpose | ||||||||||||||||||||||||||||||||||||||||||||
The procedures described below concern the preparation for analysis of final samples, sent to the control laboratories after sampling in accordance with the provisions laid down by First Commission Directive 76/371/EEC of 1 March 1976 establishing Community methods of sampling for the official control of feedingstuffs.1 | ||||||||||||||||||||||||||||||||||||||||||||
1 O.J. No. L102, 15.4.1976, p. 1. | ||||||||||||||||||||||||||||||||||||||||||||
These samples must be prepared in such a way that the amounts weighed out, as provided for in the methods of analysis, are homogeneous and representative of the final samples. | ||||||||||||||||||||||||||||||||||||||||||||
2. Precautions to be taken | ||||||||||||||||||||||||||||||||||||||||||||
All the necessary operations must be performed in such a way as to avoid as far as possible contamination of the sample and changes of its composition. Grinding, mixing and sieving should be carried out as quickly as possible with minimal exposure of the sample to the air and light. Mills and grinders likely to appreciably heat the sample should not be used. Manual grinding is recommended for feeding stuffs which are particularly sensitive to heat. Care should also be taken to ensure that the apparatus itself is not a source of contamination of trace elements. | ||||||||||||||||||||||||||||||||||||||||||||
If the preparation cannot be carried out without significant changes in the moisture content of the sample, determine the moisture content before and after preparation according to the method laid down in Part 1 of the Annex to Second Commission Directive 71/393/EEC of 18 November 1971 establishing Community methods of analysis for the official control of feedingstuffs,2 as amended by Commission Directive 73/47/EEC of 5 December 1972.3 | ||||||||||||||||||||||||||||||||||||||||||||
2 O.J. No. L279, 20.12.1971, p.7. | ||||||||||||||||||||||||||||||||||||||||||||
3 O.J. No. L83, 30.3.1973, p.35. | ||||||||||||||||||||||||||||||||||||||||||||
3. Procedure | ||||||||||||||||||||||||||||||||||||||||||||
Mix thoroughly the final sample either mechanically or manually, Divide the sample into two equal portions (the quartering method should be used where applicable). Keep one of the portions in a suitable clean, dry container, fitted with an air-tight stopper, and prepare the other portion or a representative part of it, of at least 100 g., as indicated below. | ||||||||||||||||||||||||||||||||||||||||||||
3.1 Feedingstuffs which can be ground as such | ||||||||||||||||||||||||||||||||||||||||||||
Unless otherwise specified in the methods of analysis, sieve the whole sample through a sieve with a square mesh of 1mm side (in accordance with recommendation ISO R565) after grinding, if necessary. Avoid any overgrinding. | ||||||||||||||||||||||||||||||||||||||||||||
Mix the sieved sample and collect it in a suitable clean dry container fitted with an air-tight stopper. Mix again, immediately before weighing out the amount for analysis. | ||||||||||||||||||||||||||||||||||||||||||||
3.2 Feedingstuffs which can be ground after drying | ||||||||||||||||||||||||||||||||||||||||||||
Unless otherwise specified in the methods of analysis, dry the sample to bring its moisture content down to a level of 8 to 12%, according to the preliminary drying procedure described under point 4.3 of the method of determination of moisture mentioned in section 2 above. Then proceed as indicated in section 3.1. | ||||||||||||||||||||||||||||||||||||||||||||
3.3 Liquid or semi-liquid feedingstuffs | ||||||||||||||||||||||||||||||||||||||||||||
Collect the sample in a suitable clean,dry container, fitted with an air-tight stopper. Mix thoroughly immediately before weighing out the amount for analysis. | ||||||||||||||||||||||||||||||||||||||||||||
3.4 Other feedingstuffs | ||||||||||||||||||||||||||||||||||||||||||||
Samples which cannot be prepared according to one of the above procedures should be treated by any other procedure which ensures that the amounts weighed out for the analysis are homogeneous and representative of the final samples. | ||||||||||||||||||||||||||||||||||||||||||||
4. Storage of samples | ||||||||||||||||||||||||||||||||||||||||||||
Samples must be stored at a temperature that will not alter their composition. Samples intended for the analysis of vitamins or substances which are particularly sensitive to light should be stored in brown glass containers. | ||||||||||||||||||||||||||||||||||||||||||||
B. PROVISIONS RELATING TO REAGENTS AND APPARATUS USED IN METHODS OF ANALYSIS. | ||||||||||||||||||||||||||||||||||||||||||||
1. Unless otherwise specified in the methods of analysis, all analytical reagents must be analytically pure (a.p.). When determining trace elements, the purity of the reagents must be checked by a blank test. Depending upon the results obtained, further purification of the reagents may be required. | ||||||||||||||||||||||||||||||||||||||||||||
2. Any operation involving preparation of solutions, dilution, rinsing or washing, mentioned in the methods of analysis without indication as to the nature of the solvent or diluent employed, implies that water must be used. As a general rule, water should be demineralized or distilled. In particular cases, which are indicated in the methods of analysis, it must be submitted to special procedures of purification. | ||||||||||||||||||||||||||||||||||||||||||||
3. In view of the equipment normally found in control laboratories, only those instruments and apparatus which are special or require specific usage are referred to in the methods of analysis. They must be clean especially when very small amounts of substances have to be determined. | ||||||||||||||||||||||||||||||||||||||||||||
C. APPLICATION OF METHODS OF ANALYSIS AND EXPRESSION OF THE RESULTS. | ||||||||||||||||||||||||||||||||||||||||||||
1. In general a single method of analysis is established for the determination of each substance in feedingstuffs. Where several methods are given, the particular method used by the control laboratory must be indicated on the analysis report. | ||||||||||||||||||||||||||||||||||||||||||||
2. The result given in the analysis report shall be the average value obtained from at least two determinations, carried out on separate portions of the sample, and of satisfactory repeatability. | ||||||||||||||||||||||||||||||||||||||||||||
This result shall be expressed in the manner laid down in the method of analysis to an appropriate number of significant figures and shall be corrected, if necessary, to the moisture content of the final sample prior to preparation." | ||||||||||||||||||||||||||||||||||||||||||||
and | ||||||||||||||||||||||||||||||||||||||||||||
( b ) the insertion of the following paragraphs in Part II after paragraph 27 (inserted by Regulation 2 of the Regulations of 1980): | ||||||||||||||||||||||||||||||||||||||||||||
"28. DETERMINATION OF AVOPARCIN | ||||||||||||||||||||||||||||||||||||||||||||
by diffusion in an agar medium. | ||||||||||||||||||||||||||||||||||||||||||||
1. Purpose and scope | ||||||||||||||||||||||||||||||||||||||||||||
The method is for the determination of avoparcin in feedingstuffs and premixes. The lower limit of determination is 2 mg/kg (2 ppm). The presence of polyether antibiotics may interfere in the determination. | ||||||||||||||||||||||||||||||||||||||||||||
2. Principle | ||||||||||||||||||||||||||||||||||||||||||||
The sample is extracted with a mixture of acetone/water/hydrochloric acid. The antibiotic activity of the extract is determined by measuring the diffusion of avoparcin in an agar medium inoculated with Bacillus subtilis. Diffusion is shown by the formation of zones of inhibition of the micro-organism. The diameter of these zones is taken to be in direct proportion to the logarithm of the antibiotic concentration over the range of antibiotic concentrations employed. | ||||||||||||||||||||||||||||||||||||||||||||
3. Micro-organism: Bacillus subtilis ATCC 6633 (NCIB 8054) | ||||||||||||||||||||||||||||||||||||||||||||
3.1. Maintenance of stock culture | ||||||||||||||||||||||||||||||||||||||||||||
Inoculate tubes containing slopes of culture medium (4.1.) with B. subtilis and incubate overnight at 30°C. Store the culture in a refrigerator at about 4°C. Reinoculate every month. | ||||||||||||||||||||||||||||||||||||||||||||
3.2. Preparation of the spore suspension 1 | ||||||||||||||||||||||||||||||||||||||||||||
1they give similar spore suspensions. | ||||||||||||||||||||||||||||||||||||||||||||
Harvest the growth from a recently prepared agar slope (3.1) by means of 2 to 3 ml. of sterile water. Use this suspension to inoculate 300 ml. of culture medium (4.1) contained in a Roux flask and incubate for 3-5 days at 30°C. Harvest the growth in 15 ml. of ethanol (4.2) after having checked sporulation under the microscope, and mix well. This suspension may be kept for at least 5 months at about 4°C. | ||||||||||||||||||||||||||||||||||||||||||||
4. Culture media and reagents | ||||||||||||||||||||||||||||||||||||||||||||
4.1. Culture medium 2 | ||||||||||||||||||||||||||||||||||||||||||||
2 Any commercial culture of similar composition and giving the same results may be used. | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
pH 6.5 (after sterilisation) | ||||||||||||||||||||||||||||||||||||||||||||
4.2. Ethanol 20% (v/v): dilute 200 ml of ethanol with 800 ml water | ||||||||||||||||||||||||||||||||||||||||||||
4.3. Hydrochloric acid, d: 1.18-1.19 | ||||||||||||||||||||||||||||||||||||||||||||
4.4. Sodium hydroxide, 2M solution | ||||||||||||||||||||||||||||||||||||||||||||
4.5. Phosphate buffer, 0.1M: | ||||||||||||||||||||||||||||||||||||||||||||
Potassium dihydrogen phosphate, KH2PO4: 13.6 g | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
Adjust pH to 4.5 | ||||||||||||||||||||||||||||||||||||||||||||
4.6. Mixture of acetone/water/hydrochloric acid (4.3): 65/32.5/2.5 (v/v/v) | ||||||||||||||||||||||||||||||||||||||||||||
4.7. Standard substance: avoparcin sulphate of known activity. | ||||||||||||||||||||||||||||||||||||||||||||
5. Standard solutions | ||||||||||||||||||||||||||||||||||||||||||||
Dissolve an accurately weighted quantity of approximately 10 mg of the standard substance (4.7) in phosphate buffer (4.5) and dilute with this buffer to give a stock solution containing 100µg avoparcin per ml. Stored in a stoppered flask at 4°C, this solution is stable for up to 7 days. | ||||||||||||||||||||||||||||||||||||||||||||
5.1. For premixes | ||||||||||||||||||||||||||||||||||||||||||||
From this stock solution prepare by successive dilution with buffer (4.5) the following solutions: | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
5.2 For feedingstuffs | ||||||||||||||||||||||||||||||||||||||||||||
From the stock solution prepare by successive dilution with buffer (4.5) the following solutions: | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
6. Preparation of the extract and assay solutions | ||||||||||||||||||||||||||||||||||||||||||||
6.1. Premixes | ||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest mg., a quantity of sample containing 10 to 100 mg avoparcin. Transfer to a 100 ml graduated flask with 60 ml of the mixture (4.6) and shake for 15 minutes on a mechanical shaker. Check the pH and adjust to pH 2, if necessary, with hydrochloric acid (4.3). Make up to volume with the mixture (4.6) and mix well. Filter a portion through suitable filter paper (e.g. Whatman No. 1), discarding the first 5 ml of the filtrate. Take an aliquot and adjust the pH to 4.5 with sodium hydroxide solution (4.4). Dilute this solution with buffer (4.5) to obtain an expected avoparcin concentration of 4 µg/ml (=U8). | ||||||||||||||||||||||||||||||||||||||||||||
From this solution prepare solutions U4 (expected content: 2 µg/ml, U2 (expected content: 1 µg/ml) and U1 (expected content: 0.5 ( µg/ml) by means of successive dilution (1+1) with buffer (4.5). | ||||||||||||||||||||||||||||||||||||||||||||
6.2. Feedingstuffs | ||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest mg, 50 g of sample, and place in a centrifuge tube. Add 100 ml of mixture (4.6) and shake for 30 minutes on a mechanical shaker. Clarify the extract by centrifugation (using stoppered centrifuge tubes), take an aliquot of the clarified extract (see table below) and adjust the pH to 4.5 with sodium hydroxide solution (4.4). Dilute this aliquot with buffer (4.5) to provide U8 (see table below). | ||||||||||||||||||||||||||||||||||||||||||||
From this solution prepare solutions U4 (expected content: 1.0 µg/ml), U2 (expected content: 0.5 µg/ml) and U1 (expected content: 0.25 µg/ml) by means of successive dilution (1+1) with buffer (4.5). | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
7. Assay procedure | ||||||||||||||||||||||||||||||||||||||||||||
7.1. Inoculation of the assay medium | ||||||||||||||||||||||||||||||||||||||||||||
Inoculate the assay medium (4.1) with the spore suspension (3.2) at 50-60°C. By preliminary trials on plates with assay medium (4.1) determine the quantity of spore suspension required to give the largest and clearest zones of inhibition with the various concentrations of avoparcin. | ||||||||||||||||||||||||||||||||||||||||||||
7.2. Preparation of the plates | ||||||||||||||||||||||||||||||||||||||||||||
Diffusion through agar is carried out in plates with the four concentrations of the standard solution (S8, S4, S2, S1,) and the four concentrations of the assay solution (U8, U4, U2, U1,). | ||||||||||||||||||||||||||||||||||||||||||||
These four concentrations of extract and standard must necessarily be placed in each plate. To this effect, select plates big enough to allow at least eight holes with a diameter of 10 to 13 mm and not less than 30 mm between centres to be made in the agar medium. The test may be carried out on plates consisting of a sheet of glass with a faced aluminium or plastic ring placed on top, 200 mm in diameter and 20 mm high. | ||||||||||||||||||||||||||||||||||||||||||||
Pour into the plates a quantity of the medium (4.1) inoculated as in 7.1, to give a layer about 2 mm thick (60 ml for a plate of 200 mm diameter). Allow to set in a level position, bore the holes and place in them exactly measured volumes of assay and standard solutions (between 0.10 and 0.15 ml per hole, according to the diameter). Apply each concentration at least four times so that each determination is subject to an evaluation of 32 zones of inhibition. | ||||||||||||||||||||||||||||||||||||||||||||
7.3. Incubation | ||||||||||||||||||||||||||||||||||||||||||||
Incubate the plates for 16 to 18 hours at 30°C. | ||||||||||||||||||||||||||||||||||||||||||||
8. Evaluation | ||||||||||||||||||||||||||||||||||||||||||||
Measure the diameter of the zones on inhibition to the nearest 0.1 mm. Record the mean measurements for each concentration of semi-logarithmic graph paper showing the logarithm of the concentrations in relation to the diameters of the zones of inhibition. Plot the 'best fit' lines of both the standard solution and the extract, for example as below. Determine the 'best fit' point for the standard highest level (SL) using the formula: | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
Determine the 'best fit' point for the standard highest level (SH) using the formula: | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
Similarly calculate the 'best fit' points for the extract lowest level (UL) and the extract highest level (UH) by substituting U1, U2, U4 and U8 for S1, S2, S4 and S8 in the above formulae. | ||||||||||||||||||||||||||||||||||||||||||||
Record the calculated SL and SH values on the same graph paper and join them to give the 'best fit' line for the standard solution. Similarly record UL and UH and join them to give the 'best fit' line for the extract. | ||||||||||||||||||||||||||||||||||||||||||||
In the absence of any interference the lines should be parallel. For practical purposes the lines can be considered parallel if the values (SH-SL) and (UH-UL) do not differ by more than 10% from their mean value. | ||||||||||||||||||||||||||||||||||||||||||||
If the lines are found to be non-parallel either U1 and S1 or U8 and S8 may be discarded and SL, SH, UL and UH calculated, using the alternative formulae, to give alternative 'best fit' lines: | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
and similarly for UL and UH. The alternative 'best fit' lines should be checked for parallelism as before. The fact that the result has been calculated from three levels should be noted on the final report. | ||||||||||||||||||||||||||||||||||||||||||||
When the lines are considered as being parallel calculate the logarithm of the relative activity (log. A) by means of one of the following formulae: | ||||||||||||||||||||||||||||||||||||||||||||
For 4 levels | ||||||||||||||||||||||||||||||||||||||||||||
(c) Log. A= | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
For 3 levels | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
Real activity = supposed activity x relative activity. | ||||||||||||||||||||||||||||||||||||||||||||
If the relative activity is found to be outside the range of 0.5 to 2.0, then repeat the assay making appropriate adjustments to the extract concentrations or, if this is not possible, to the standard solutions. When the relative activity cannot be brought into the required range, any result obtained must be considered as approximate and this should be noted on the final report. | ||||||||||||||||||||||||||||||||||||||||||||
When the lines are considered as not being parallel, repeat the determination. If parallelism is still not achieved, the determination must be considered as unsatisfactory. | ||||||||||||||||||||||||||||||||||||||||||||
9. Repeatability | ||||||||||||||||||||||||||||||||||||||||||||
The difference between the results of two determinations carried out on the same sample by the same analyst should not exceed: | ||||||||||||||||||||||||||||||||||||||||||||
—2 mg/kg, in absolute value, for contents of avoparcin from 2 and up to 10 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||
—20% of the higher result for contents of 10 to 25 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||
—5 mg/kg, in absolute value, for contents of 25 to 50 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||
—10% of the higher result for contents above 50 mg/kg. | ||||||||||||||||||||||||||||||||||||||||||||
29. DETERMINATION OF MONENSIN SODIUM | ||||||||||||||||||||||||||||||||||||||||||||
by diffusion in an agar medium | ||||||||||||||||||||||||||||||||||||||||||||
1. Purpose and scope | ||||||||||||||||||||||||||||||||||||||||||||
The method is for the determination of monensin sodium in feedingstuffs and premixes. The lower limit of determination is 10 mg/kg (10 ppm).1 | ||||||||||||||||||||||||||||||||||||||||||||
1 1 mg monesin sodium is equivalent to 1000 UK units. | ||||||||||||||||||||||||||||||||||||||||||||
2. Principle | ||||||||||||||||||||||||||||||||||||||||||||
The sample is extracted with 90% methanol. The extract is submitted to appropriate procedures according to the monensin sodium content of the sample. The antibiotic activity is determined by measuring the diffusion of monensin sodium in an agar medium inoculated with Bacillus subtilis. Diffusion is shown by the formation of zones of inhibition of the micro-organism. The diameter of these zones is taken to be in direct proportion to the logarithm of the antibiotic concentration over the range of antibiotic concentrations employed. The sensitivity of this assay system is reduced in the presence of sodium ions. | ||||||||||||||||||||||||||||||||||||||||||||
3. Micro-organism: Bacillus subtilis ATCC 6633 (NCIB 8054) | ||||||||||||||||||||||||||||||||||||||||||||
3.1. Maintenance of stock culture | ||||||||||||||||||||||||||||||||||||||||||||
Inoculate tubes containing slopes of culture medium (4.1) with B. subtilis and incubate overnight at 30°C. Store the culture in a refrigerator at about 4°C. Reinoculate every month. | ||||||||||||||||||||||||||||||||||||||||||||
3.2. Preparation of the spore suspension 1 | ||||||||||||||||||||||||||||||||||||||||||||
1 Other methods may be used provided that it has been established that they give similar spore suspensions. | ||||||||||||||||||||||||||||||||||||||||||||
Harvest the growth from a recently prepared agar slope (3.1) by means of 2 to 3 ml of sterile water. Use this suspension to inoculate 300 ml of culture medium (4.1) contained in a Roux flask and incubate for 3-5 days at 30°C. Harvest the growth in 15 ml of 20% ethanol (4.3), after having checked sporulation under the microscope and mix well. This suspension may be kept for at least 5 months at about 4°C. | ||||||||||||||||||||||||||||||||||||||||||||
4. Culture media and reagents | ||||||||||||||||||||||||||||||||||||||||||||
4.1. Culture medium2 | ||||||||||||||||||||||||||||||||||||||||||||
2 Any commercial culture medium of similar composition and giving the same results may be used. | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
pH 6.5 (after sterilisation) | ||||||||||||||||||||||||||||||||||||||||||||
4.2. Assay medium | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
4.3. Ethanol 20% (v/v): dilute 200 ml of ethanol with 800 ml water. | ||||||||||||||||||||||||||||||||||||||||||||
4.4. Methanol, anhydrous. | ||||||||||||||||||||||||||||||||||||||||||||
4.5. Methanol 90% (v/v): dilute 900 ml of methanol (4.4) with 100 ml water. | ||||||||||||||||||||||||||||||||||||||||||||
4.6. Methanol 50% (v/v): dilute 500 ml of methanol (4.4) with 500 ml water. | ||||||||||||||||||||||||||||||||||||||||||||
4.7. Aluminium oxide, granulated (Alcoa F, 20 mesh; Activated Alumina UGI: F Lancaster and Co., or equivalent). | ||||||||||||||||||||||||||||||||||||||||||||
4.8. Standard substances: monensin sodium of known activity (e.g. from International Laboratory for Biological Standards, Central Veterinary Laboratory, Weybridge, Surrey KT15 3NB). | ||||||||||||||||||||||||||||||||||||||||||||
5. Apparatus | ||||||||||||||||||||||||||||||||||||||||||||
5.1. Rotary vacuum evaporator, with a 250 ml round-bottom flask. | ||||||||||||||||||||||||||||||||||||||||||||
5.2. Glass tube for chromatography, internal diameter: approximately 25 mm; length: approximately 400 mm, with an open end of approximately 2 mm diameter. | ||||||||||||||||||||||||||||||||||||||||||||
5.3.Glass tube for chromatography, internal diameter: approximately 11 mm; length: approximately 300 mm, with an open end of approximately 2 mm diameter. | ||||||||||||||||||||||||||||||||||||||||||||
6. Standard solutions | ||||||||||||||||||||||||||||||||||||||||||||
Dissolve an accurately weighed quantity of the standard substance (4.8) in methanol (4.4) and dilute to give a stock solution containing 800 µg monensin sodium per ml. Stored in stoppered flasks at 4°C, this solution is stable for up to two weeks. | ||||||||||||||||||||||||||||||||||||||||||||
From this stock solution prepare by successive dilution with 50% methanol (4.6) the following solutions: | ||||||||||||||||||||||||||||||||||||||||||||
S8 8.0 µg/ml | ||||||||||||||||||||||||||||||||||||||||||||
S4 4.0 µg/ml | ||||||||||||||||||||||||||||||||||||||||||||
S2 2.0 µg/ml | ||||||||||||||||||||||||||||||||||||||||||||
S1 1.0 µg/ml | ||||||||||||||||||||||||||||||||||||||||||||
7. Preparation of the extract | ||||||||||||||||||||||||||||||||||||||||||||
7.1. Extraction | ||||||||||||||||||||||||||||||||||||||||||||
7.1.1. Premixes | ||||||||||||||||||||||||||||||||||||||||||||
Weigh to the nearest mg, 2 g of sample and place in a centrifuge tube. Add 100 ml of 90% methanol (4.5), shake and centrifuge for a few minutes. Dilute the supernatant solution with 50% methanol (4.6) to obtain an expected monensin sodium content of 8 µg/ml (=U8). | ||||||||||||||||||||||||||||||||||||||||||||
7.1.2. Feedingstuffs with a level of monensin sodium not lower than 50 ppm | ||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest mg, a quantity of sample between 10 and 20 g and place in a conical flask. Add 100 ml of 90% methanol (4.5), shake for 15 minutes and leave to settle. | ||||||||||||||||||||||||||||||||||||||||||||
Insert a cotton-wool plug at the narrow end of a glass tube (5.2), add aluminium oxide (4.7) with gentle tapping until the aluminium oxide reaches a height of 75 to 80 mm. | ||||||||||||||||||||||||||||||||||||||||||||
Decant the extract on to the aluminium oxide column and collect the filtrate. Dilute 30 ml of the filtrate to 50 ml with water. Make subsequent dilutions with 50% methanol (4.6) to obtain an expected monensin sodium content of 8 µg/ml (=U8). | ||||||||||||||||||||||||||||||||||||||||||||
7.1.3. Feedingstuffs with a level of monensin sodium lower than 50 ppm (up to the limit of 10 ppm) | ||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest mg, a quantity of sample between 10 and 20 g and place in a centrifuge tube. Add 100 ml of 90% methanol (4.5) and shake for 15 minutes. Centrifuge till clear. | ||||||||||||||||||||||||||||||||||||||||||||
If the sample contains 20 ppm of monensin sodium take 40 ml of the supernatant liquid. If it contains 10 ppm take 80 ml and evaporate to dryness under vacuum on a rotary evaporator (5.1) at not more than 40°C. Dissolve the residue in 10 ml of 90% methanol (4.5). | ||||||||||||||||||||||||||||||||||||||||||||
Insert a cotton-wool plug at the narrow end of a glass tube (5.3), add aluminium oxide (4.7) with gentle tapping until the aluminium oxide reaches a height of 75 to 80 mm. | ||||||||||||||||||||||||||||||||||||||||||||
Decant the methanolic solution on to the aluminium oxide column and collect the filtrate. Wash the column with 10 ml of 90% methanol (4.5) and combine the washings with the filtrate. | ||||||||||||||||||||||||||||||||||||||||||||
Evaporate the solution to dryness under vacuum on a rotary evaporator (5.1) at less than 40°C. Dissolve the residue in 10 ml of anhydrous methanol (4.4) and make up to 20 ml with water. Centrifuge the solution at, at least, 400 r/min for at least 5 minutes. Make subsequent dilutions with 50% methanol (4.6) to obtain an expected monensin sodium content of 8 µg/ml (=U8). | ||||||||||||||||||||||||||||||||||||||||||||
7.2. Assay solutions | ||||||||||||||||||||||||||||||||||||||||||||
From solution U8 prepare solutions U4 (expected content: 4 µg/ml), U2 (expected content: 2 µg/ml) and U1 (expected content: 1 µg/ml) by means of successive dilution (1 + 1) with 50% methanol (4.6). | ||||||||||||||||||||||||||||||||||||||||||||
8. Assay procedure | ||||||||||||||||||||||||||||||||||||||||||||
8.1. Inoculation of the assay medium | ||||||||||||||||||||||||||||||||||||||||||||
Inoculate the assay medium (4.2) with the spore suspension (3.2) at 50-60°C. By preliminary trials on plates with assay medium (4.2) determine the quantity of spore suspension required to give the largest and clearest zones of inhibition with the various concentrations of monensin sodium. | ||||||||||||||||||||||||||||||||||||||||||||
8.2. Preparation of the plates | ||||||||||||||||||||||||||||||||||||||||||||
Diffusion through agar is carried out in plates with the four concentrations of the standard solution (S8, S4, S2, S1) and the four concentrations of the assay solution (U8, U4, U2, U1). | ||||||||||||||||||||||||||||||||||||||||||||
These four concentrations of extract and standard must necessarily be placed in each plate. To this effect, select plates large enough to allow at least eight holes with a diameter of 10 to 13 mm and not less than 30 mm between centres to be made in the agar medium. The test may be carried out on plates consisting of a sheet of glass with a faced aluminium or plastic ring placed on top, 200 mm in diameter and 20 mm high. | ||||||||||||||||||||||||||||||||||||||||||||
Pour into the plates a quantity of the medium (4.2) inoculated as in 8.1., to give a layer about 2 mm thick (60 ml for a plate of 200 mm diameter). Allow to set in a level position, bore the holes and place in them exactly measured volumes of assay and standard solutions (between 0.10 and 0.15 ml per hole, according to the diameter). Apply each concentration at least four times so that each determination is subject to an evaluation of 32 zones of inhibition. | ||||||||||||||||||||||||||||||||||||||||||||
8.3. Incubation | ||||||||||||||||||||||||||||||||||||||||||||
Incubate the plates for approximately 18 hours at 35-37°C. | ||||||||||||||||||||||||||||||||||||||||||||
9. Evaluation | ||||||||||||||||||||||||||||||||||||||||||||
Measure the diameter of the zones of inhibition to the nearest 0.1 mm. Record the mean measurements for each concentration on semi-logarithmic graph paper showing the logarithm of the concentrations in relation to the diameters of the zones of inhibition. Plot the 'best fit' lines of both the standard solution and the extract, for example as below. | ||||||||||||||||||||||||||||||||||||||||||||
Determine the 'best fit' point for the standard lowest level (SL) using the formula: | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
Determine the 'best fit' point for the standard highest level (SH) using the formula: | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
Similarly calculate the 'best fit' points for the extract lowest level (UL) and the extract highest level (UH) by substituting U1, U2, U4 and U8 for S1, S2, S4 and S8 in the above formulae. | ||||||||||||||||||||||||||||||||||||||||||||
Record the calculated SL and SH values on the same graph paper and join them to give the 'best fit' line for the standard solution. Similarly record UL and UH and join them to give the 'best fit' line for the extract. | ||||||||||||||||||||||||||||||||||||||||||||
In the absence of any interference the lines should be parallel. For practical purposes the lines can be considered parallel if the values (SH-SL) and (UH-UL) do not differ by more than 10% from their mean value. | ||||||||||||||||||||||||||||||||||||||||||||
If the lines are found to be non-parallel either U1 and S1 or U8 and S8 may be discarded and SL, SH, UL and UH calculated, using the alternative formulae, to give alternative 'best fit' lines: | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
and similarly for UL and UH. The alternative 'best fit' lines should be checked for parallelism as before. The fact that the result has been calculated from three levels should be noted on the final report. | ||||||||||||||||||||||||||||||||||||||||||||
When the lines are considered as being parallel, calculate the logarithm of the relative activity (Log. A) by means of one of the following formulae: | ||||||||||||||||||||||||||||||||||||||||||||
For 4 levels | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
For 3 levels | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
or | ||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||
A | ||||||||||||||||||||||||||||||||||||||||||||
Real activity = supposed activity x relative activity. | ||||||||||||||||||||||||||||||||||||||||||||
If the relative activity is found to be outside the range of 0.5 to 2.0 then repeat the assay making appropriate adjustments to the extract concentrations, or, if this is not possible, to the standard solutions. When the relative activity cannot be brought into the required range, any result obtained must be considered as approximate and this should be noted on the final report. | ||||||||||||||||||||||||||||||||||||||||||||
When the lines are considered as not being parallel, repeat the determination. If parallelism is still not achieved, the determination must be considered as unsatisfactory. | ||||||||||||||||||||||||||||||||||||||||||||
10. Repeatability | ||||||||||||||||||||||||||||||||||||||||||||
The difference between the results of two determinations carried out on the same sample by the same analyst should not exceed: | ||||||||||||||||||||||||||||||||||||||||||||
—20% of the higher result for contents of 10 to 25 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||
—5 mg/kg, in absolute value, for contents of 25 to 50 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||
—10% of the higher result for contents above 50 mg/kg." | ||||||||||||||||||||||||||||||||||||||||||||
GIVEN under my Official Seal this 4th day of August, 1982. | ||||||||||||||||||||||||||||||||||||||||||||
BRIAN LENIHAN, | ||||||||||||||||||||||||||||||||||||||||||||
Minister for Agriculture. | ||||||||||||||||||||||||||||||||||||||||||||
EXPLANATORY NOTE. | ||||||||||||||||||||||||||||||||||||||||||||
These Regulations, which implement the provisions of Commission Directives 81/680/EEC and 81/715/EEC prescribe: | ||||||||||||||||||||||||||||||||||||||||||||
( a ) general provisions governing the analysis of feeding stuffs; | ||||||||||||||||||||||||||||||||||||||||||||
( b ) methods by which analysis of animal feeding stuffs for avoparcin and monensin sodium are to be carried our for the purposes of the European Communities (Feeding Stuffs) (Additives) Regulations, 1974 to 1981. |