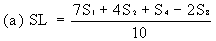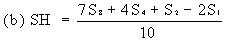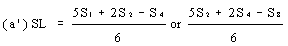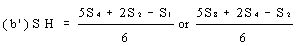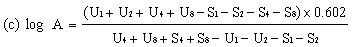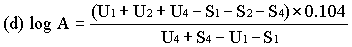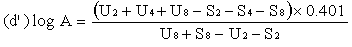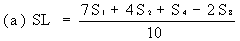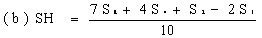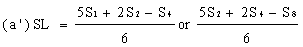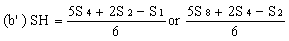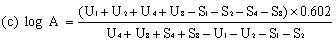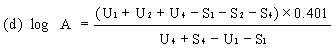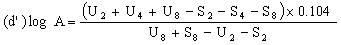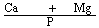S.I. No. 14/1980 - European Communities (Feeding Stuffs) (Methods of Analysis (Amendment) and Methods of Sampling) Regulations, 1980.
S.I. No. 14 of 1980. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
EUROPEAN COMMUNITIES (FEEDING STUFFS) (METHODS OF ANALYSIS (AMENDMENT) AND METHODS OF SAMPLING) REGULATIONS, 1980. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
I, RAY MacSHARRY, Minister for Agriculture, in exercise of the powers conferred on me by section 3 of the European Communities Act, 1972 (No. 27 of 1972), for the purpose of giving effect to Commission Directive No. 76/371/EEC of 1 March, 1976, and Commission Directive No. 78/633/EEC of 15 June, 1978, hereby make the following regulations: 1. (1) These Regulations may be cited as the European Communities (Feeding Stuffs) (Methods of Analysis (Amendment) and Methods of Sampling) Regulations, 1980. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(2) These Regulations shall come into operation on the 18th day of February, 1980. 2. The European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1978 ( S.I. No. 250 of 1978 ), are hereby amended— | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(a) by the addition of the following to Part II: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
25. DETERMINATION OF ZINC BACITRACIN BY DIFFUSION IN AN AGAR MEDIUM | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
1. PURPOSE AND SCOPE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The method is for the determination of zinc bacitracin in feedingstuffs, concentrates and premixes. The lower limit of determination is 5 mg/kg (5 ppm). (a). The method is not valid in the presence of interfering substances, particularly large amounts of copper and ascorbic acid. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
2. PRINCIPLE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The sample is extracted at pH 2 with a mixture of methanol/water/hydrochloric acid. The extract is brought to pH 6.5, concentrated (where necessary) and diluted. Its antibiotic activity is determined by measuring the diffusion of zinc bacitracin in an agar medium inoculated with Micrococcus luteus (Syn. M. flavus). Diffusion is shown by the formation of zones of inhibition of the micro-organism. The diameter of these zones is taken to be in direct proportion to the logarithm of the antibiotic concentration over the range of antibiotic concentrations employed. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(a) 1 mg feed grade zinc bacitracin is equivalent to 42 international units (i.u.). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3. MICRO-ORGANISM: MICROCOCCUS LUTEUS AT CC 10240 (NCTC 7743) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.1 Maintenance of stock culture | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Inoculate Micrococcus luteus on to agar slopes of culture medium (4.1). Incubate for 24 hours at 30 to 37°C, store in a refrigerator at about 4°C and reinoculate every two weeks on to agar slopes. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.2 Preparation of the bacterial suspension (a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Harvest the growth from a recently prepared agar slope (3.1) with 2 to 3 m1 of sodium chloride solution (4.3). Use this suspension to inoculate 250 m1 of culture medium (4.1) contained in a Roux flask and incubate for 18 to 20 hours at 30 to 37°C. Harvest the growth in 25 m1 of sodium chloride solution (4.3) and mix. Dilute the suspension to one-tenth with sodium chloride solution (4.3). The light transmission of the suspension must be about 75% measured at 650 nm in a 1 cm cell against sodium chloride solution (4.3). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4. CULTURE MEDIA AND REAGENTS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.1 Culture medium (b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.2 Assay medium (b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(a) Other methods may be used provided that it has been established that they give similar bacterial suspensions. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(b) Any commercial culture medium of similar composition and giving the same results may be used. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.3 Sodium chloride solution 0.8% (w/v): dissolve 8g of sodium chloride in water and dilute to 1000ml; sterilise. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.4 Mixture of methanol (pure) /water/hydrochloric acid (d: 1.18 to 1.19): 80/17. 5/2. 5 (v/v/v). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.5 Phosphate buffer, pH 6.5. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Potassium hydrogen phosphate K2HPO4 (22.15g) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Potassium dihydrogen phosphate KH2PO4 (27.85g) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Water to 1,000 ml. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.6 Hydrochloric acid (d: 1.18 to 1.19) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.7 Hydrochloric acid (0.1 N). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.8 Sodium hydroxide 1 N solution. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.9 Bromocresol purple solution 0.04% (w/v): dissolve 0.1g of bromocresol purple in 18.5 ml of 0.01 N sodium hydroxide solution. Make up the volume to 250 ml with water and mix. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.10 Standard substance: zinc bacitracin of known activity (in i.u.). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5. Standard solutions | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Weigh out a quantity of standard zinc bacitracin (4.10) corresponding to 1050 i.u. (according to the activity indicated). Add 5 ml of 0.1 N hydrochloric acid (4.7) and leave to stand for 15 minutes. Add 30 ml of water, adjust the pH to 4.5 with phosphate buffer pH 6.5 (4.5) (about 4 ml), make up to a volume of 50 ml with water and mix well (1 ml=21 i.u.). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
From this solution prepare by successive dilution with phosphate buffer pH 6.5 (4.5) the following solutions: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
S8 0.42 i.u./ml | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
S4 0.21 i.u./ml | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
S2 0.105 i.u./ml | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
S1 0.0525 i.u./ml | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6. PREPARATION OF THE EXTRACT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6.1 Extraction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6.1.1 Concentrates, premixes and mineral feeds | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest mg, a quantity of sample containing 2.0 to 5.0g, add 30 ml of the mixture (4.4) and shake briefly. Check that tha pH is about 2. Shake for 10 minutes, add 30 m1 of phosphate buffer pH 6.5 (4.5) and shake for 15 minutes. Dilute with phosphate buffer (4.5) to obtain an expected zinc bacitracin content of 0.42 i.u./ml (=U8). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6.1.2 Protein concentrates | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest mg, 10 g of sample, add 50 m1 of the mixture (4.4) and shake briefly. Check that the pH is about 2. Shake for 10 minutes, add 50 ml of phosphate buffer pH 6.5 (4.5) and shake for 15 minutes. Dilute with phosphate buffer (4.5) to obtain an expected zinc bacitracin content of 0.42 i.u./ml (=U8). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6.1.3 Other feeds | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest mg, 10g of sample (20.0g for an expected zinc bacitracin content of 5 mg/kg), add 25 ml of the mixture (4.4) and shake for about 10 minutes. Add 25 ml of phosphate buffer pH 6.5 (4.5), shake for 15 minutes and centrifuge. Take 20 m1 of the supernatant solution and adjust the pH to 6.5 by means of 1 N sodium hydroxide solution (4.8) with the bromocresol purple solution (4.9) as indicator. Evaporate to about 4 m1 in a rotary evaporator at a temperature not exceeding 35°C. Dilute the residue with water to obtain an expected zinc bacitracin content of 0.42 i.u./ml (= U8). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6.2 Assay solutions | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
From solution U8 prepare solutions U4 (expected content: 0.21 i.u./ml), U2 (expected content: 0.105 i.u./ml) and U1 (expected content: 0.0525 i.u./ml) by means of successive dilution (1+1) with phosphate buffer (4.5). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7. Assay Procedure | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7.1 Inoculation of the assay medium | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Inoculate the assay medium (4.2) with the bacterial suspension (3.2) at about 50°C. By preliminary trials on plates with assay medium (4.2) determine the quantity of bacterial suspension required to give the largest and clearest zones of inhibition with the various concentrations of zinc bacitracin. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7.2 Preparation of the plates | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Diffusion through agar is carried out in plates with concentrations of the standard solution (S8, S4, S2, S1) and the four concentrations of the assay solution (U8, U4, U2, U1). These four concentrations of extract and standard must necessarily be placed on each plate. To this effect select plates big enough to allow at least eight holes with a diameter of 10 to 13 mm and not less than 30 mm between centres to be made in the agar medium. The test may be carried out on plates consisting of a sheet of glass with a faced aluminium or plastic ring placed on top, 200 mm in diameter and 20 mm high. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Pour into the plates a quantity of the medium (4.2), inoculated as in point 7.1, to give a layer about 2 mm thick (50 ml for a plate of 200 mm diameter). Allow to set in a level position, bore the holes and place in them exactly measured volumes of assay and standard solutions (between 0.10 and 0.15 ml per hole, according to the diameter). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Apply each concentration at least four times so that each determination is subject to an evaluation of 32 zones of inhibition. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7.3 Incubation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Incubate the plates for 16 to 18 hours at 28 to 30°C. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
8. Evalation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Measure the diameter of the zones of inhibition to the nearest 0.1 mm. Record the mean measurements for each concentration on semi-logarithmic graph paper showing the logarithm of the concentrations in relation to the diameters of the zones of inhibition. Plot the 'best fit' lines of both the standard solution and the extract, for example as below. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Determine the 'best fit' point for the standard lowest level (SL) using the formula: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Determine the 'best fit' point for the standard highest level (SH) using the formula: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Similarly calculate the 'best fit' points for extract lowest level (UL) and the extract highest level (UH) by substituting U1, U2, U4 and U8 for S1, S2, S4 and S8 in the above formulae. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Record the calculated SL and SH values on the same graph paper and join them to give the 'best fit' line for the standard solution. Similarly record UL and UH and join them to give the 'best fit' line for the extract. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In the absence of any interference the lines should be parallel. For practical purposes the lines can be considered parallel if the values (SH-SL) and (UH-UL) do not differ by more than 10% from their mean value. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
If the lines are found to be non-parallel, either U1, and S1 or U8 and S8 may be discarded and SL, SH, UL and UH calculated, using the alternative formulae, to give alternative 'best fit' lines: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
and similarly for UL and UH. The alternative 'best fit' lines should be checked for parallelism as before. The fact that the result has been calculated from three levels should be noted on the final report. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
When the lines are considered as being parallel, calculate the logarithm of the relative activity (log A) by means of one of the following formulae. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For four levels | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For three levels | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
or | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Real activity = supposed activity x relative activity. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
When the lines are considered as not being parallel, repeat the determination. If parallelism is still not achieved, calculate the logarithm of the relative activity (log A) by means of formula (c). The result obtained must however be considered as approximate and this should be noted in the final report. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
9. Repeatability | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The difference between the results of two determinations carried out on the same sample by the same analyst should not exceed: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
20% of the higher result for contents of zinc bacitracin equal to or greater than 5 but not greater than 25 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5 mg/kg, in absolute value, for contents greater than 25 but not greater than 50 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
10% of the higher result value for contents greater than 50 mg/kg. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
26. DETERMINATION OF FLAVOPHOSPHOLIPOL BY DIFFUSION IN AN AGAR MEDIUM. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
1. Purpose and Scope | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The method is for the determination of flavophospholipol in feedingstuffs, concentrates and premixes. The lower limit of determination is 1 mg/kg (1ppm). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
2. Principle | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The sample is extracted with diluted methanol by heating under reflux. After centrifuging, the extract is purified (where necessary) by treatment with ion exchange resins and diluted. Its antibiotic activity is determined by measuring the diffusion of flavophospholipol in an agar medium inoculated with Staphylococcus aureus. Diffusion is shown by the formation of zones of inhibition of the micro-organism. The diameter of these zones is taken to be in direct proportion to the logarithm of the antibiotic concentration over the range of antibiotic concentrations employed. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3. MICRO-ORGANISM: STAPHYLOCOCCUS AUREUS AT CC 6538 P(NCIB 8625). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.1 Maintenance of stock culture. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Inoculate Staphylococcus aureus on to agar slopes of culture medium (4.1). Incubate for 24 hours at 37°C, store in a refrigerator at about 4°C and reinoculate every month on to agar slopes. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.2 Preparation of the bacterial suspension (a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Set aside two tubes containing the stock culture (3.1) and reinoculate them weekly. Incubate for 24 hours at 37°C and store in a refrigerator at about 4°C. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
24 hours before the assay, inoculate with this growth two to four tubes containing slopes of culture medium (4.1), Incubate for 16 to 18 hours at 37°C. Make a suspension of the growth in the sodium chloride solution (4.3). The light transmission of the suspension must be about 40%, measured at 578 nm in a 1cm cell against sodium chloride solution (4.3). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4. Culture Media and Reagents | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.1 Culture medium (b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.2 Assay medium | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.2.2 Seed layer | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
As for point 4.1, with the addition of 2.0g of silicone anti-foaming emulsion. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.3 Sidium chloride solution 0.4% (w/v): dissolve 4g of sodium chloride in water and dilute to 1000 ml; sterilise. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(a) Other methods may be used provided that it has been established that they give similar bacterial suspensions. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(b) Any commercial culture medium of similar composition and giving the same results may be used, e.g. oxoid antibiotic medium 1 (CM 327) with an addition of oxoid agar No. 3 (L 13). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(c) Any commercial culture medium of similar composition and giving the same results may be used, e.g. oxoid antibiotic medium 2 (CM 335) with an addition of oxoid agar No. 3 (L13). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.4 Methanol (pure). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.5 Methanol 50% (v/v): dilute 500 ml of methanol (4.4) with 500 ml of water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.6 Methanol 80% (v/v): dilute 800 ml of methanol (4.4) with 200 ml of water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.7 Tris (hydroxymethyl) aminomethane. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.8 Potassium chloride methanolic solution 1.5% (w/v): dissolve 1.5g of potassium chloride in 20 ml of water, make up the volume to 100 ml with methanol (4.4). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.9 Cation exchanger: Dowex 50W x 8, 20 to 50 mesh, Na form (cat. Serva No. 41600) or equivalent. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.10 Anion exchanger: Dowex 1 x 2, 50 to 100 mesh, C1 form (cat. Serva No. 41010) or equivalent. Before use, keep for 12 to 14 hours in 80% methanol (4.6). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.11 Glass wool. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.12 pH indicator paper (pH 6.6 to 8.1). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.13 Ascorbic acid. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.14 Standard substance: flavophospholipol of known activity. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5. Apparatus | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.1 Glass tube for chromatography, internal diameter approximately 9mm, length; 150 to 200 mm, fitted with a stopcock at the narrowed part of the lower end and a ground-glass joint (to connect with the dropping funnel (5.2) at the upper end). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.2 Dropping funnel fitted with a stopcock and a ground-glass joint. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.3 Conical flask with ground-glass joint. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.4 Reflux condenser with ground-glass joint. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6. Standard solutions | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Dissolve an accurately weighed quantity of the standard substance (4.14) in 50% methanol (4.5) and dilute to give a stock solution containing 100 µg flavophospholipol per millilitre. Stored in stoppered flasks at 4°C this solution is stable for up to two months. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
From this stock solution prepared by successive dilution with 50% methanol (4.5) the following solutions: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
S8 0.2 µg/ml | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
S4 0.1 µg/ml | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
S2 0.05 µg/ml | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
S1 0.025 µg/ml | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7. Preparation of the Extract | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7.1 Extraction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7.1.1 Concentrates, premixes and mineral feeds | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Weigh out a quantity of sample of 2.0 to 5.0 g and add about 150 mg of ascorbic acid (4.13). Homogenise with 150 ml of 50% mehtanol (4.5) in a conical flask (5.3) and adjust the pH to 8.1 to 8.2 with about 400 mg of tris (hydroxymethyl) aminomethane (4.7). Check the pH with indicator paper (4.12). Allow to stand for 15 minutes, then readjust the pH to 8.1 to 8.2 with tris (hydroxymethyl) aminomethane (4.7) and boil for 10 minutes under reflux (5.4) with constant stirring. Allow to cool, centrifuge the mixture and decant the extract. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7.1.2 Other feeds | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Weigh, to the nearest mg, between 5 to 30 g of sample containing at least 30 µg of flavophospholipol. Mix with 150 ml of 50% methanol (4.5) in a conical flask (5.3) and adjust the pH to 8.1 to 8.2 with about 400 mg of tris (hydroxymethyl) aminomethane (4.7). Check the pH with indicator paper (4.12). Allow to stand for 15 minutes, then readjust the pH to 8.1 to 8.2 with tris (hydroxymethyl) aminomethane (4.7) and boil under reflux (5.4) for 10 minutes with constant stirring. Allow to cool, centrifuge the mixture and decant the extract. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7.2 Purification (this step may be omitted for concentrates, premixes and mineral feeds) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Mix 110 ml of the extract with 11g of the cation exchanger (4.9), boil under reflux (5.4) for one minute with constant stirring. Separate the cation exchanger by centrifugation or filtration. Mix 100 ml of the extract with 150 ml of methanol (4.4) and store the solution for 12 to 15 hours at 4°C. Filter off the flocculent mass while cold. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Insert a glass wool plug (4.11) at the bottom end of a glass tube (5.1), pour into the tube 5 ml of the anion exchanger (4.10) and wash the column with 100 ml of 80% methanol (4.6). Using the funnel (5.2), transfer to the column a volume of filtrate of at least 100 ml which is expected to contain 16 µg of flavophospholipol (200 ml for a 30 g sample of feedingstuff at 1 mg/kg). Where necessary, before application to the column, dilute the filtrate with 80% methanol (4.6) to obtain an expected flavophospholipol content of 16 µg/100 ml. Adjust the flow rate to about 2 ml/minute. Discard the effluent. Then wash the column with 50 ml of 80% methanol (4.6) and discard the effluent. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Elute the flavophospholipol with the methanolic solution of potassium chloride (4.8) keeping the flow rate to about 2ml/minute. Collect 50 ml of the eluate in a graduated flask, add 30 ml of water and mix. This solution should have a flavophospholipol content of 0.2 µg/ml (=U8). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7.3 Assay solutions | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Where necessary (i.e. when the purification step has been ommitted), dilute the extract obtained in point 7.1.1 with 50% methanol (4.5) to obtain an expected flavophospholipol content of 0.2 µg/ml (=U8). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
From solution U8 prepare solutions U4 (expected content: 0.1 µg/ml), U2 (expected content: 0.05 µg/ml) and U1 (expected content: 0.025 µg/ml) by means of successive dilution (1+1) with 50% methanol (4.5). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
8. Assay Procedure | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
8.1 Inoculation of the assay medium | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Inoculate the assay medium (4.2.2) with the bacterial suspension (3.2) at about 50°C. By preliminary trials on plates with assay medium (4.2.2) determine the quantity of bacterial suspension required to give the largest and clearest zones of inhibition with the various concentrations of flavophospholipol (about 30 ml/litre). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
8.2 Preparation of the plates | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Diffusion through agar is carried out in plates with the four concentrations of the standard solution (S8, S4, S2, S1) and the four concentrations of the assay solution (U8, U4, U2, U1). These for concentrations of extract and standard must necessarily be placed in each plate. To this effect, select plates big enough to allow at least eight holes with a diameter of 10 to 13 mm and not less than 30 mm between centres to be made in the agar medium. The test may be carried out on plates consisting of a sheet of glass with a faced aluminium or plastic ring placed on top, 200 mm in diameter and 20 mm high. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Pour into the plates a quantity of the medium (4.2.1) to give a layer about 1.5 mm thick (45 ml for a plate of 200 mm diameter). Allow to set in a level position and then over-layer with a quantity of the medium (4.2.2) inoculated as in point 8.1 to give a layer 1 mm thick (30 ml for a plate of 200 mm diameter). Allow to set again in a level position, bore the holes and place in them exactly measured volumes of assay and standard solutions (between 0.10 and 0.15 ml per hole according to the diameter). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Apply each concentration at least four times so that each determination is subject to an evaluation of 32 zones of inhibition. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
8.3 Incubation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Incubate the plates for 16 to 18 hours at 28 to 30°C. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
9. Evaluation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Measure the diameter of the zones of inhibition to the nearest 0.1 mm. Record the mean measurements for each concentration on semi-logarithmic graph paper showing the logarithm of the concentrations in relation to the diameters of the zones of inhibition. Plot the best fit lines of both the standard solution and the extract, for example as below. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Determine the 'best fit' point for the standard lowest level (SL) using the formula: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Similarly calculate the 'best fit' points for the extract lowest level (UL) and the extract highest level (UH) by substituting U1, U2, U4 and U8 for S1, S2, S4 and S8 in the above formulae. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Record the calculated SL and SH values on the same graph paper and join them to give the 'best fit' line for the standard solution. Similarly record UL and UH and join them to give the 'best fit' line for the extract. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In the absence of any interference the lines should be parallel. For practical purposes the lines can be considered parallel if the values (SH-SL) and (UH-UL) do not differ by more than 10% from their mean value. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
If the lines are found to be non-parallel, either U1 and S1 or U8 and S8 may be discarded and SL, SH, UL and UH calculated, using the alternative formulae, to give alternative 'best fit' lines. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
and similarly for UL and UH. The alternative 'best fit' lines should be checked for parallelism as before. The fact that the result has been calculated from three levels should be noted on the final report. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
When the lines are considered as being parallel, calculate the logarithm of the relative activity (log A) by means of one of the following formulae. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For four levels | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For three levels | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
or | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Real activity =supposed activity x relative activity. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
When the lines are considered as not being parallel, repeat the determination. If parallelism is still not achieved, calculate the logarithm of the relative activity (log A) by means of formula (c). The result obtained must however be considered as approximate and this should be noted in the final report. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
10. Repeatability | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The difference between the results of two determinations carried out on the same sample by the same analyst should not exceed: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
0.5 mg/kg, in absolute value, for contents of flavophospholipol equal to or greater than 1 but not greater than 2 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
25% related to the highest value for contents greater than 2 but not greater than 10 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
20% related to the highest value for contents greater than 10 but not greater than 25 mg/kg: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5 mg/kg, in absolute value, for contents greater than 25 but not greater than 50 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
10% related to the highest value for contents greater than 50 mg/kg. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
27. DETERMINATION OF THE TRACE ELEMENTS IRON, COPPER, MANGANESE AND ZINC | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
1. Purpose and scope | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The method is for the determination of the trace elements iron, copper, manganese and zinc in feedingstuffs. The lower limits of determination are:— | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
2. Principle | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The sample, or the residue resulting from ashing, if there is organic matter present, is treated with hydrochloric acid. The elements iron, copper, manganese and zinc are determined, after appropriate dilution, by atomic absorption spectrometry. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3. Reagents | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Introductory comments | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For preparation of the reagents and analytical solutions use water free from the cations to be determined, obtained either by double distilling water in a borosilicate glass or quartz still or by double treatment on ion exchange resin. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The reagents must be of at least analytical grade. Freedom from the element to be determined must be checked in a blank experiment. If necessary, the reagents must be further purified. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In determining trace elements it is important to be alert to the risks of contamination, particularly by zinc, copper and iron. For this reason, the equipment used in preparing the samples must be free of these metals. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
To reduce the general risk of contamination, work in a dust-free atmosphere with scrupulously clean equipment and carefully washed glassware. The determination of zinc is particularly sensitive to many types of contamination e.g. from glassware, reagents, dust, etc. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In place of the standard solutions described below, commercial standard solutions may be used provided that they are guaranteed and have been checked before use. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.1 Hydrochloric acid (d: 1.18) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.2 Hydrochloric acid (6 N). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.3 Hydrochloric acid (0.5 N). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.4 Hydrofluoric acid 38 to 40% (v/v) having an iron content of less than 1 mg Fe/litre and a residue after evaporation of less than 10 mg (as sulphate)/litre. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.5 Sulphuric acid (d: 1.84) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.6 Hydrogen peroxide (approximately 100 volumes of oxygen (30% by weight)). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.7 Standard iron solution (1,000 µg Fe/ml) prepared as follows: dissolve 1 g of iron wire in 200 ml of 6 N hydrochloric acid (3.2), add 16 ml of hydrogen peroxide (3.6) and make up to one litre with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.7.1 Working standard iron solution (100 µg Fe(ml) prepared by diluting the standard solution (3.7) 1 + 9 with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.8 Standard copper solution (1,000 µg Cu/ml) prepared as follows: dissolve 1 g of copper in powder form in 25 ml of 6 N hydrochloric acid (3.2), add 5 ml hydrogen peroxide (3.6) and make up to one litre with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.8.1 Working standard copper solution (10 µg Cu/ml) prepared by diluting the standard solution (3.8) 1 + 9 with water and then diluting the resulting solution 1 + 9 with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.9 Standard manganese solution (1,000 µg Mn/ml) prepared as follows: dissolve 1 g of manganese in powder form in 25 ml of 6 N hydrochloric acid (3.2) and make up to one litre with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.9.1 Working standard manganese solution (10 µg Mn/ml) prepared by diluting the standard solution (3.9)1 + 9 with water and then diluting the resulting solution 1 + 9 with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.10 Standard zinc solution (1,000 µg Zn/ml) prepared as follows: dissolve 1 g of zinc in strip or leaf form in 25 ml of 6 N hydrochloric acid (3.2) and make up to one litre with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.10.1 Working standard zinc solution (10 µg Zn/ml) prepared by diluting the standard solution (3.10) 1 + 9 with water and then diluting the resulting solution 1 + 9 with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.11 Lanthanum chloride solution prepared as follows: dissolve 12 g of lanthanum oxide in 150 ml of water, add 100 ml of 6 N hydrochloric acid (3.2) and make up to one litre with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4. Apparatus | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.1 Muffle furnace with temperature regulator and recorder. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.2 Glassware must be of resistant borosilicate type and it is recommended to use apparatus which is reserved exclusively for trace element determination. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.3 Platinum crucible and (optional) quartz crucible. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4.4 Atomic absorption spectrophotometer meeting the requirements of the method with regard to sensitivity and precision in the required range. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5. Procedure | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.1 Samples containing organic matter. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.1.1 Ashing and preparation of the solution for analysis (* ). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(*) Green fodder (fresh or dried) is liable to contain large amounts of vegetable silica, which may retain trace elements and must be removed. For samples of these feedingstuffs, therefore, the following modified procedure must be followed. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Carry out operation 5.1.1. (I) as far as the filtration. Wash the filter paper containing the insoluble residue twice with boiling water and place it in a platinum crucible (4.3). Ignite in the muffle furnace (4.1) at a temperature below 550°C until all carbonaceous material has completely disappeared. Allow to cool, add a few drops of water followed by 10 to 15 ml of hydrofluoric acid (3.4) and evaporate to dryness at about 150°C. If any silica remains in the residue, redissolve it in a few millilitres of hydrofluoric acid (3.4) and evaporate to dryness. Add five drops of sulphuric acid (3.5) and heat until no more white fumes are given off. After the addition of 5 ml of 6 N hydrochloric acid (3.2) and about 30 ml of water, heat, filter the solution into the 250 ml volumetric flask and make up to the mark with water (HC l concentration about 0.5 N). Proceed then with the determination from point 5.1.3. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(I) Place 5 to 10 g of sample weighed to the nearest 0.2 mg in a quartz or platinum crucible (4.3) (see Note (a)), dry in an oven at 105°C and introduce the crucible into the cold muffle furnace (4.1). Close the furnace (see Note (b)) and gradually raise the temperature to 450 to 475°C over about 90 minutes. Maintain this temperature for 4 to 16 hours (e.g. overnight) to remove carbonaceous material and then open the furnace and allow to cool (see Note (c)). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Wash the crucible out with a total of about 5 ml of hydrochloric acid (3.1) and add the latter slowly and carefully to the beaker (there may be a vigorous reaction due to CO2 formation). Add hydrochloric acid (3.1) dropwise with agitation until all effervescence has stopped. Evaporate to dryness, occasionally stirring with a glass rod. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Next add 15 ml of 6 N hydrochloric acid (3.2) to the residue followed by about 120 ml of water. Stir with the glass rod, which should be left in the beaker, and cover the beaker with a watchglass. Bring gently to the boil and maintain at boiling point until no more ash can be seen to dissolve. Filter on ash-free filter paper and collect the filtrate in a 250 ml volumetric flask. Wash the beaker and filter with 5 ml of hot 6 N hydrochloric acid (3.2) and twice with boiling water. Fill the volumetric flask up to the mark with water (HC l concentration about 0.5N). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(II) If the residue in the filter appears black (carbon), put it back in the furnace and ash again at 450 to 475°C. This ashing, which only requires a few hours (about three to five hours), is complete when the ash appears white or nearly white. Dissolve the residue with about 2 ml of hydrochloric acid (3.1), evaporate to dryness and add 5 ml of 6 N hydrochloric acid (3.2). Heat, filter the solution into the volumetric flask and make up to the mark with water (HC l concentration about 0.5 N). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Notes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(a) The weight of sample to be ashed is calculated from the approximate trace element content of the feedingstuff in relation to the sensitivity of the spectrophotometer used. For certain feedingstuffs low in trace elements it may be necessary to start with a 10 to 20 g sample and make up the final solution to only 100 ml. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(b) Ashing must be carried out in a closed furnace without injection of air or oxygen. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(c) The temperature indicated by the pyrometer must not exceed 475°C. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.1.2 Spectrophotometric determination | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.1.2.1 Preparation of calibration solutions | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For each of the elements to be determined, prepare from the working standard solutions given in points 3.7.1., 3.8.1., 3.9.1. and 3.10.1. a range of calibration solutions, each calibration solution having a HC l concentration of about 0.5 N and (in the cases of iron, manganese and zinc) a lanthanum chloride concentration equivalent to 0.1% La /w/v). The trace element concentrations selected must lie within the range of sensitivity of the spectrophotometer used. The tables below show, by way of example, the compositions of typical ranges of calibration solutions; depending, however, on the type and sensitivity of spectrophotometer used it may be necessary to select other concentrations. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Iron | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
+ 10 ml of lanthanum chloride solution (3. 11) and make up to 100 ml with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Copper | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Make up to 100 ml with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Manganese | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
+ 10 ml lanthanum chloride solution (3.11) and make up to 100 ml with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Zinc | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
+10 ml lanthanum chloride solution (3.11) and make up to 100 ml with water. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.1.2.2 Proparation of solution for analysis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the determination of copper, the solution prepared from point 5.1.1. can normally be used directly. If necessary to bring its concentration within the range of the calibration solutions, an aliquot portion may be pipetted into a 100 ml. volumetric flask and made up to the mark with 0.5 N hydrochloric acid (3.3). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the determination of iron, manganese and zinc, pipette an aliquot portion off the solution prepared from point 5.1.1 into a 100 ml volumetric flask, add 10 ml of lanthanum chloride solution (3.11) and make up to the mark with 0.5 N hydrochloric acid (3.3) (see also point 8 'Observation'). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.1.2.3 Blank experiment | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The blank experiment must include all the prescribed steps of the procedure except that the sample material is omitted. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The calibration solution 'O' must not be used as the blank. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.1.2.4 Measurement of the atomic absorption | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Measure the atomic absorption of the calibration solutions and of the solution to be analysed using an oxidising air-acetylene flame at the following wavelengths. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Carry out each measurement four times. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5.2 Mineral Feedingstuffs | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
If the sample contains no organic matter, prior ashing is unnecessary. Proceed as described in point 5.1.1 ( I ) starting from the second paragraph. Evaporation with hydrofluoric acid may be omitted. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6. CALCULATION OF RESULTS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Using a calibration curve, calculate the trace element concentration in the solution to be analysed and expressed the result in milligrams of trace element per kilogram of sample (ppm). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7. REPEATABILITY | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The difference between the results of two parallel determinations carried out on the same sample by the same analyst should not exceed: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
— 5 mg/kg, in absolute value, for contents of the trace element concerned not greater than 50 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
— 10% of the higher result for contents of the trace element concerned greater than 50 but not greater than 100 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
— 10 mg/kg, in absolute value, for contents of the trace element concerned greater than 100 but not greater than 200 mg/kg; | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
— 5% of the higher result for contents of the trace element concerned greater than 200 mg/kg. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
8. OBSERVATION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The presence of large quantities of phosphates may interfere with the determination of iron, manganese and zinc. Such interference must be corrected by addition of lanthanum chloride solution (3.11). If, however, in the sample the weight ratio | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
is greater than 2, addition of lanthanum chloride solution (3.11) to the solution for analysis and to the calibration solutions may be omitted". | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
( b ) by the substitution of the following for the Schedule thereto: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
"SCHEDULE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
This is to certify that the above-mentioned sample, which was duly fastened and sealed, has been analysed under the direction of the *State Chemist/*Assistant State Chemist and that the result of the analysis is as follows:— | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
This certificate is given under the European Communities (Feeding Stuffs) (Methods of Analysis) Regulations, 1978. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3. (1) The manner in which samples shall be taken and dealt with for the purposes of either the European Communities (Feeding Stuffs) (Additives) Regulations, 1974 and 1979, or the European Communities (Feeding Stuffs) (Tolerances of Undesirable Substances and Products) Regulations, 1977 ( S.I. No. 246 of 1977 ), shall be according to the methods for quality and composition described in the Annex to Commission Directive No. 76/371/EEC of 1 March 1976(a). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(a)OJ No. L 102/1 15.4.1976 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(2) Paragraph (1) of this Regulation is in addition to and not in substitution for Regulation 6(4) of the European Communities (Feeding Stuffs) (Tolerances of Undesirable Substances and Products) Regulations, 1977. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
GIVEN under my Official Seal, this 17th day of January, 1980. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
RAY MacSHARRY, | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Minister for Agriculture. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
EXPLANATORY NOTE. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
These Regulations, which implement certain of the provisions of Commission Directives 76/371/EEC and 78/633/EEC adopted pursuant to Council Directive 70/373/EEC of the 20th July, 1970 prescribe:— | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
( a ) the manner in which samples of animal feeding stuffs shall be taken and dealt with for the purposes of the European Communities (Feeding Stuffs) (Additives) Regulations, 1974 and 1979 ( S.I. No. 302 of 1974 and S.I. No. 6 of 1979 ) and the European Communities (Feeding Stuffs) (Tolerances of Undesirable Substances and Products) Regulations, 1977 ( S.I. No. 246 of 1977 ); | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
( b ) Further methods by which analysis of animal feeding stuffs is to be carried out for the purposes of the European Communities (Feeding Stuffs) (Additives) Regulations, 1974 and 1979. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||