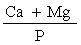S.I. No. 13/1980 - Feeding Stuffs and Mineral Mixtures (Methods of Sampling) and Fertilisers, Feeding Stuffs and Mineral Fixtures (Methods of Analysis (Amendment)) Regulations, 1980.
S.I. No. 13 of 1980. | ||||||||||||||||||||||||||
FEEDING STUFFS AND MINERAL MIXTURES (METHODS OF SAMPLING) AND FERTILISERS, FEEDING STUFFS AND MINERAL FIXTURES (METHODS OF ANALYSIS (AMENDMENT)) REGULATIONS, 1980. | ||||||||||||||||||||||||||
I, RAY MacSHARRY, Minister for Agriculture, in exercise of the powers conferred on me by sections 8 and 11 of the Fertilisers, Feeding Stuffs and Mineral Mixtures Act, 1955 (No. 8 of 1955), hereby make the following regulations: 1. (1) These Regulations may be cited as the Feeding Stuffs and Mineral Mixtures (Methods of Sampling) and Fertilisers, Feeding Stuffs and Mineral Mixtures (Methods of Analysis (Amendment)) Regulations, 1980. | ||||||||||||||||||||||||||
(2) These Regulations shall come into force on the 18th day of February, 1980. 2. In these Regulations "the Principal Regulations" means the Fertilisers, Feeding Stuffs and Mineral Mixtures Regulations, 1957 ( S.I. No. 264 of 1957 ). 3. Article 7 (1) of the Principal Regulations shall have effect in relation to feeding stuffs as if for "as specified in the subsequent paragraphs of this Article" there were substituted "according to the methods for quality and composition described in the Annex to Commission Directive No. 76/371/EEC of 1 March, 1976"(a). | ||||||||||||||||||||||||||
(a) OJ No. L 102/1. 15.4.1976. 4. Part I of the Schedule to the Fertilisers, Feeding Stuffs and Mineral Mixtures (Methods of Analysis) Regulations, 1978 ( S.I. No. 249 of 1978 ), is hereby amended by the substitution of the following paragraphs for paragraphs 19, 20 and 21: | ||||||||||||||||||||||||||
"DETERMINATION OF THE TRACE ELEMENTS IRON, COPPER AND MANGANESE | ||||||||||||||||||||||||||
1. Purpose and Scope | ||||||||||||||||||||||||||
The method is for the determination of the trace elements iron, copper and manganese in feedingstuffs. The lower limits of determination are:— | ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
2. Principle | ||||||||||||||||||||||||||
The sample, or the residue resulting from ashing, if there is organic matter present, is treated with hydrochloric acid. The elements iron, copper and manganese are determined, after appropriate dilution, by atomic absorption spectrometry. | ||||||||||||||||||||||||||
3. Reagents | ||||||||||||||||||||||||||
Introductory comments | ||||||||||||||||||||||||||
For preparation of the reagents and analytical solutions use water free from the cations to be determined, obtained either by double distilling water in a borosilicate glass or quartz still or by double treatment on ion exchange resin. | ||||||||||||||||||||||||||
The reagents must be of at least analytical grade. Freedom from the element to be determined must be checked in a blank experiment. If necessary, the reagents must be further purified. | ||||||||||||||||||||||||||
In determining trace elements it is important to be alert to the risks of contamination, particularly, by zinc, copper and iron. For this reason, the equipment used in preparing the samples must be free of these metals. | ||||||||||||||||||||||||||
To reduce the general risk of contamination, work in a dust-free atmosphere with scrupulously clean equipment and carefully washed glassware. | ||||||||||||||||||||||||||
In place of the standard solutions described below, commercial standard solutions may be used provided that they are guaranteed and have been checked before use. | ||||||||||||||||||||||||||
3.1. Hydrochloric acid (d:1.18). | ||||||||||||||||||||||||||
3.2. Hydrochloric acid (6N). | ||||||||||||||||||||||||||
3.3. Hydrochloric acid (0.5N). | ||||||||||||||||||||||||||
3.4. Hydrofluoric acid 38 to 40% (v/v) having an iron content of less than 1 mg Fe/litre and a residue after evaporation of less than 10 mg (as sulphate)/litre. | ||||||||||||||||||||||||||
3.5. Sulphuric acid (d:1.84). | ||||||||||||||||||||||||||
3.6. Hydrogen peroxide (approximately 100 volumes of oxygen (30% by weight)). | ||||||||||||||||||||||||||
3.7. Standard iron solution (1,000 µg Fe/ml) prepared as follows: dissolve 1 g of iron wire in 200 ml of 6 N hydrochloric acid (3.2), add 16 ml of hydrogen peroxide (3.6) and make up to one litre with water. | ||||||||||||||||||||||||||
3.7.1. Working standard iron solution (100 µg Fe/ml) prepared by diluting the standard solution (3.7) 1+9 with water. | ||||||||||||||||||||||||||
3.8. Standard copper solution (1,000 µg Cu/ml) prepared as follows: dissolve 1 g of copper in powder form in 25 ml of 6 N hydrochloric acid (3.2), add 5 ml hydrogen peroxide (3.6) and make up to one litre with water. | ||||||||||||||||||||||||||
3.8.1. Working standard copper solution (10 µg Cu/ml) prepared by diluting the standard solution (3.8) 1+9 with water and then diluting the resulting solution 1+9 with water. | ||||||||||||||||||||||||||
3.9. Standard manganese solution (1,000 µg Mn/ml) prepared as follows: dissolve 1 g of manganese in powder form in 25 ml of 6 N hydrochloric acid (3.2) and make up to one litre with water. | ||||||||||||||||||||||||||
3.9.1. Working standard manganese solution (10 µg Mn/ml) prepared by diluting the standard solution (3.9) 1+9 with water and then diluting the resulting solution 1+9 with water. | ||||||||||||||||||||||||||
3.10. Lanthanum chloride solution prepared as follows: dissolve 12 g of lanthanum oxide in 150 ml of water, add 100 ml of 6 N hydrochloric acid (3.2) and make up to one litre with water. | ||||||||||||||||||||||||||
4. Apparatus | ||||||||||||||||||||||||||
4.1. Muffle furnace with temperature regulator and recorder. | ||||||||||||||||||||||||||
4.2. Glassware must be of resistant borosilicate type and it is recommended to use apparatus which is reserved exclusively for trace element determinations. | ||||||||||||||||||||||||||
4.3. Platinum crucible and (optional) quartz crucible. | ||||||||||||||||||||||||||
4.4. Atomic absorption spectrophotometer meeting the requirements of the method with regard to sensitivity and precision in the required range. | ||||||||||||||||||||||||||
5. Procedure | ||||||||||||||||||||||||||
5.1. Samples containing organic matter | ||||||||||||||||||||||||||
5.1.1. Ashing and preparation of the solution for analysis* | ||||||||||||||||||||||||||
*Green fodder (fresh or dried) is liable to contain large amounts of vegetable silica, which may retain trace elements and must be removed. For samples of these feedingstuffs, therefore, the following modified procedure must be followed. | ||||||||||||||||||||||||||
Carry out operation 5.1.1. (I) as far as the filtration. Wash the filter paper containing the insoluble residue twice with boiling water and place it in a platinum crucible (4.3). Ignite in the muffle furnace (4.1) at a temperature below 550°C until all carbonaceous material has completely disappeared. Allow to cool, add a few drops of water followed by 10 to 15 ml of hydrofluoric acid (3.4) and evaporate to dryness at about 150°C. If any silica remains in the residue, redissolve it in a few millilitres of hydrofluoric acid (3.4) and evaporate to dryness. Add five drops of sulphuric acid (3.5) and heat until no more white fumes are given off. After the addition of 5 ml of 6 N hydrochloric acid (3.2) and about 30 ml of water, heat, filter the solution into the 250 ml volumetric flask and make up to the mark with water (HC1 concentration about 0·5 N). Proceed then with the determination from point 5.1.3. | ||||||||||||||||||||||||||
(I) Place 5 to 10 g of sample weighed to the nearest 0.2 mg in a quartz or platinum crucible (4.3) (see Note (a)), dry in an oven at 105°C and introduce the crucible into the cold muffle furnace (4.1). Close the furnace (see Note (b)) and gradually raise the temperature to 450 to 475°C over about 90 minutes. Maintain this temperature for 4 to 16 hours (e.g. overnight) to remove carbonaceous material and then open the furnace and allow to cool (see Note (c)). | ||||||||||||||||||||||||||
Wash the crucible out with a total of about 5 ml of hydrochloric acid (3.1) and add the latter slowly and carefully to the beaker (there may be a vigorous reaction due to CO2 formation). Add hydrochloric acid (3.1) dropwise with agitation until all effervescence has stopped. Evaporate to dryness, occasionally stirring with a glass rod. | ||||||||||||||||||||||||||
Next add 15 ml of 6 N hydrochloric acid (3.2) to the residue followed by about 120 ml of water. Stir with the glass rod, which should be left in the beaker, and cover the beaker with a watchglass. Bring gently to the boil and maintain at boiling point until no more ash can be seen to dissolve. Filter on ash-free filter paper and collect the filtrate in a 250 ml volumetric flask. Wash the beaker and filter with 5 ml of hot 6 N hydrochloric acid (3.2) and twice with boiling water. Fill the volumetric flask up to the mark with water (HC1 concentration about 0.5 N). | ||||||||||||||||||||||||||
(II) If the residue in the filter appears black (carbon), put it back in the furnace and ash again at 450 to 475°C. This ashing, which only requires a few hours (about three to five hours), is complete when the ash appears white or nearly white. Dissolve the residue with about 2 ml of hydrochloric acid (3.1), evaporate to dryness and add 5 ml of 6 N hydrochloric acid (3.2). Heat, filter the solution into the volumetric flask and make up to the mark with water (HC1 concentration about 0.5 N). | ||||||||||||||||||||||||||
Note: | ||||||||||||||||||||||||||
( a ) The weight of sample to be ashed is calculated from the approximate trace element content of the feedingstuff in relation to the sensitivity of the spectrophotometer used. For certain feedingstuffs low in trace elements it may be necessary to start with a 10 to 20 g sample and make up the final solution to only 100 ml. | ||||||||||||||||||||||||||
( b ) Ashing must be carried out in a closed furnace without injection of air or oxygen. | ||||||||||||||||||||||||||
( c ) The temperature indicated by the pyrometer must not exceed 475°C. | ||||||||||||||||||||||||||
5.1.2. Spectrophotometric determination | ||||||||||||||||||||||||||
5.1.2.1. Preparation of calibration solutions | ||||||||||||||||||||||||||
For each of the elements to be determined, prepare from the working standard solutions given in points 3.7.1., 3.8.1., 3.9.1. and 3.10.1. a range of calibration solutions, each calibration solution having an HC1 concentration of about 0.5 N and (in the cases of iron and manganese) a lanthanum chloride concentration equivalent to 0.1% La (w/v). The trace element concentrations selected must lie within the range of sensitivity of the spectrophotometer used. The tables below show, by way of example, the compositions of typical ranges of calibration solutions; depending, however, on the type and sensitivity of spectrophotometer used it may be necessary to select other concentrations. | ||||||||||||||||||||||||||
Iron | ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
+10 ml of lanthanum chloride solution (3.11) and make up to 100 ml with water. | ||||||||||||||||||||||||||
Copper. | ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
Make up to 100 ml with water. | ||||||||||||||||||||||||||
Manganese. | ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
+10 ml lanthanum chloride solution (3.11) and make up to 100 ml with water. | ||||||||||||||||||||||||||
5.1.2.2. Preparation of solution for analysis | ||||||||||||||||||||||||||
For the determination of copper, the solution prepared from point 5.1.1. can normally be used directly. If necessary to bring its concentration within the range of the calibration solutions, an aliquot portion may be pipetted into a 100 ml volumetric flask and made up to the mark with 0.5 N hydrochloric acid (3.3). | ||||||||||||||||||||||||||
For the determination of iron and manganese, pipette an aliquot portion of the solution prepared from point 5.1.1. into a 100 ml volumetric flask, add 10 ml of lanthanum chloride solution (3.11) and make up to the mark with 0.5 N hydrochloric acid (3.3) (see also point 8 "Observation"). | ||||||||||||||||||||||||||
5.1.2.3. Blank experiment | ||||||||||||||||||||||||||
The blank experiment must include all the prescribed steps of the procedure except that the sample material is omitted. | ||||||||||||||||||||||||||
The calibration solution "0" must not be used as the blank. | ||||||||||||||||||||||||||
5.1.2.4. Measurement of the atomic absorption | ||||||||||||||||||||||||||
Measure the atomic absorption of the calibration solutions and of the solution to be analysed using an oxidising air-acetylene flame at the following wavelengths: | ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
Carry out each measurement four times. | ||||||||||||||||||||||||||
5.2 Mineral feedingstuffs | ||||||||||||||||||||||||||
If the sample contains no organic matter, prior ashing is unnecessary. Proceed as described in point 5.1.1. (I) starting from the second paragraph. Evaporation with hydrofluoric acid may be omitted. | ||||||||||||||||||||||||||
6. Calculation of Results | ||||||||||||||||||||||||||
Using a calibration curve, calculate the trace element concentration in the solution to be analysed and express the result in milligrams of trace element per kilogram of sample (ppm). | ||||||||||||||||||||||||||
7. Repeatability | ||||||||||||||||||||||||||
The difference between the results of two parallel determinations carried out on the same sample by the same analyst should not exceed: | ||||||||||||||||||||||||||
— 5 mg/kg, in absolute value, for contents of the trace element concerned not greater than 50 mg/kg; | ||||||||||||||||||||||||||
— 10% of the higher result for contents of the trace element concerned greater than 50 but not greater than 100 mg/kg; | ||||||||||||||||||||||||||
— 10 mg/kg, in absolute value, for contents of the trace element concerned greater than 100 but not greater than 200 mg/kg; | ||||||||||||||||||||||||||
— 5% of the higher result for contents of the trace element concerned greater than 200 mg/kg. | ||||||||||||||||||||||||||
8. Observation | ||||||||||||||||||||||||||
The presence of large quantities of phosphates may interfere with the determination of iron and manganese. Such interference must be corrected by addition of lanthanum chloride solution (3.11). If, however, in the sample the weight ratio | ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
is greater than 2, addition of lanthanum chloride solution (3.11) to the solution for analysis and to the calibration solutions may be ommitted", and the Principal Regulations shall be construed and have effect accordingly. | ||||||||||||||||||||||||||
GIVEN under my Official Seal this 17th day of January, 1980. | ||||||||||||||||||||||||||
RAY MacSHARRY, | ||||||||||||||||||||||||||
Minister for Agriculture. | ||||||||||||||||||||||||||
EXPLANATORY NOTE. | ||||||||||||||||||||||||||
These Regulations amend (a) the provisions of Article 7 of the Fertilisers, Feeding Stuffs and Mineral Mixtures Regulations, 1957 ( S.I. No. 264 of 1957 ) on the manner in which samples of animal feeding stuffs are to be taken and dealt with, and (b) the provisions of the Fertilisers, Feeding Stuffs and Mineral Mixtures (Methods of Analysis) Regulations, 1978 ( S.I. No. 249 of 1978 ), so as to give statutory effect to certain of the provisions of Commission Directives 76/371/EEC and 78/633/EEC, adopted pursuant to Council Directive 70/373/EEC of the 20th July 1970. |