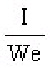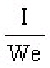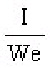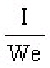S.I. No. 365/1934 - The Therapeutic Substances (Saorstát Eireann) Regulations, 1934.
THE THERAPEUTIC SUBSTANCES (SAORSTÁT EIREANN) REGULATIONS, 1934. | ||||
The Minister for Local Government and Public Health in exercise of the powers conferred on him by the Therapeutic Substances Act, 1932 , does by this his order after consultation with the Therapeutic Substances Advisory Committee make the following Regulations, that is to say:— | ||||
|
PART I.GENERAL. | ||||
|
1 Interpretation. | 1.—(1) These Regulations may be cited as the Therapeutic Substances (Saorstát Eireann) Regulations, 1934. | |||
(2) The provisions relating to labelling other than that contained in paragraph (e) of Article 8 (1) of these Regulations shall come into operation on the 1st day of March, 1935, but save as aforesaid these Regulations shall come into operation on the 1st day of June, 1935. | ||||
|
2 .. | 2.—(1) In these Regulations, unless the context otherwise requires— | |||
"the Act" means the Therapeutic Substances Act, 1932 ; | ||||
"the Minister" means the Minister for Local Government and Public Health; | ||||
"therapeutic substance" or "substance" means one of the therapeutic substances specified in the Schedule to the Act, or such other substance or substances as may from time to time be declared by any Order of the Minister for the time being in force to be therapeutic substances to which the Act applies. | ||||
(2) For the purposes of these Regulations the millilitre may be used wherever the cubic centimetre is indicated. | ||||
(3) The Interpretation Act, 1923 , applies to the interpretation of these Regulations, as it applies to the interpretation of an Act of the Oireachtas. | ||||
|
3 Licences and Applications for Licences. | 3.—(1) Such of the forms set out in the First Schedule to these Regulations as is applicable to the case (or a form substantially to the like effect) shall be used whenever a licence under these Regulations is granted by the Minister or an application for such a licence is made to the Minister. | |||
(2) Licences granted under these Regulations shall, unless sooner suspended or revoked under the Act, continue in force for a period of two years from the date thereof. | ||||
|
PART II.LICENCES FOR MANUFACTURE OF THERAPEUTIC SUBSTANCES. | ||||
|
4 .. | 4.—Before a licence to manufacture a therapeutic substance for sale (in this Part of these Regulations referred to as "a manufacturer's licence") is issued, the applicant shall satisfy the Minister that upon the issue of the licence the conditions set out in Article 5 of these Regulations will be observed. | |||
|
5 .. | 5.—Every manufacturer's licence shall be subject to the special conditions, if any, set out in the Schedule to these Regulations which relates to the therapeutic substance in respect of which the licence is granted (in these Regulations referred to as "the relative Schedule"), and to the following general conditions:— | |||
(a) the licensee shall provide and maintain an adequate staff and adequate premises and plant for the proper manufacture of the substance in respect of which the licence is issued; | ||||
(b) the licensee shall either— | ||||
(i) provide and maintain an adequate staff and adequate premises and plant for carrying out such tests of the strength, quality and purity of the substance as, pursuant to Part IV of these Regulations, may be required to be carried out by him, including proper housing for animals used for the purpose of such tests, or | ||||
(ii) make arrangements with some institution approved by the Minister for such tests to be regularly carried out on his behalf by that institution; | ||||
(c) the licensee shall not manufacture for sale the therapeutic substance in respect of which the licence is granted elsewhere than on the premises specified in the licence; | ||||
(d) the licensee shall keep permanent records of the details of manufacture of each batch of the substance which is issued for sale and of the application of the tests thereto in such form as to be available for inspection and to be easily identified by reference to the number of the batch as shown on the label of each container; | ||||
(e) the licensee shall from time to time report to the Minister any changes in the expert staff responsible for the manufacture or testing of the substance and any material alterations in the premises or plant used for that purpose which have been made since the date of the last inspection made on behalf of the licensing authority before the issue of the licence; | ||||
(f) the licensee shall on request furnish to the Minister from every batch of the substance or from such batch or batches as the Minister may from time to time specify a sample of such amount as the Minister may consider adequate for any examination required to be made; and the licensee shall, if so required, furnish full protocols of the tests which have been applied; | ||||
(g) if the Minister so directs, the licensee shall not sell or offer for sale any batch in respect of which a sample is or protocols are furnished under the last preceding sub-paragraph until a certificate authorizing the sale of the batch has been issued to him by or on behalf of the Minister; | ||||
(h) the licensee shall, on being informed by the Minister that any part of any batch of the substance has been found by the Minister not to conform with the standards of strength, quality or purity specified in these. Regulations and on being directed so to do, withdraw the remainder of that batch from sale and, so far as may in the particular circumstances of the case be practicable, recall all issues already made from that batch; | ||||
(i) the licensee shall comply with the provisions of Parts III and IV of these Regulations, and of the Schedules referred to therein, and with such further requirements, if any, as may be specified in any regulations which may be made under the Act, and of which the Minister has given the licensee not less than one month's notice. | ||||
|
PART III.PROVISIONS WITH REGARD TO THE NAMES OF THERAPEUTIC SUBSTANCES AND TO CONTAINERS, LABELS, ETC. | ||||
|
6 Name of Substance. | 6.—If any therapeutic substance is advertised or sold as a proprietary medicine or is contained in a medicine so advertised or sold, the name stated in the relative Schedule as being the accepted scientific name or name descriptive of the true nature and origin (hereinafter referred to as the "proper name") of the substance shall appear on the label in the manner prescribed in this Part of these Regulations. | |||
|
7 Containers. | 7.—(1) No therapeutic substance intended for injection shall be sold or offered for sale unless it has been sealed in a previously sterilized glass container in such manner as will in the opinion of the Minister suffice to preclude the access of bacteria: | |||
(2) When any therapeutic substance is issued in liquid form in containers which are sealed in such a manner that portions of the contents can be withdrawn for use on different occasions, the liquid shall contain a sufficient proportion of some antiseptic to prevent the growth of any organism which may be accidentally introduced in the process of removing a portion of the contents of the container. | ||||
(3) The container shall comply with such further requirements, if any, as are specified in the relative Schedule. | ||||
(4) The Minister may in the case of any particular preparation of any therapeutic substance dispense with any of the requirements of this Article or of the relative Schedule, and may make such additional requirements as, having regard to the nature of the preparation, he may deem necessary. | ||||
|
8 Labelling. | 8.—(1) Every phial, ampoule or other sealed container of a therapeutic substance shall bear a label on which is printed or written in indelible ink the following particulars and such further particulars, if any, as may be specified in the relative Schedule:— | |||
(a) the proper name of the substance in letters not less conspicuous than those in which the proprietary name, if any, is printed or written, and following immediately after or under such proprietary name; | ||||
(b) the number of every licence under which the substance or any of its constituents is manufactured or if imported, the name of the manufacturer; | ||||
(c) a distinctive batch number, that is to say, the number by reference to which the prescribed tests and details of manufacture of the particular batch from which the substance in the container is taken are permanently recorded and available for inspection; | ||||
(d) where a test for potency in units is required by these Regulations in respect of a substance other than vaccine lymph a statement of the potency in units defined in terms of relation to the standard preparation specified in the relative Schedule: | ||||
(e) if the substance is intended to be used solely for veterinary purposes, the words "to be used solely for veterinary purposes." | ||||
(2) The particulars prescribed in sub-paragraphs (a), (b) and (c) of the preceding paragraph shall be printed or written in indelible ink either on the label borne by a container of vaccine lymph or on a label or wrapper affixed to any package in which the container is issued for sale. | ||||
(3) The following particulars and such further particulars, if any, as may be specified in the relative Schedule shall be printed or written in indelible ink either on the label borne by the container of any therapeutic substance or on a label or wrapper affixed to any package in which any such container is issued for sale:— | ||||
(a) the name and address of the manufacturer of the final product; | ||||
(b) the number of the manufacture or import licence; | ||||
(c) the date on which the manufacture of the particular batch from which the substance in the container is taken was completed, as defined in the relative Schedule, or if there is no definition in the relative Schedule as hereafter defined in this Article; | ||||
(d) where a test for maximum toxicity is required by these Regulations, a statement that the substance has passed such test; | ||||
(e) where a test for potency or maximum toxicity is required, the date up to which the substance, if kept under suitable conditions, may be expected to retain a potency not less than that stated on the label to the container, or not to acquire a toxicity greater than that permitted by the test, as the case may be; | ||||
(f) where an antiseptic substance has been added, the nature and the percentage proportion introduced; | ||||
(g) the precautions necessary for preserving the properties of the contents to the date indicated in sub-paragraph (e) of this paragraph. | ||||
(4) For the purposes of sub-paragraph (c) of the last preceding paragraph the date on which the manufacture of a batch is completed shall be— | ||||
(a) in cases where a test for potency or toxicity is required by these Regulations, or not being so required, is accepted by the Minister as sufficient for the purpose of fixing the date of completion of manufacture, the date on which the test was completed, or the date on which the substance was removed from cold storage after having there been kept at a temperature not exceeding 5°C. continuously for a period not exceeding two years from the time when the last test was completed; | ||||
(b) in cases where no such test is required or accepted— | ||||
(i) if the substance is a serum obtained from living animals, the earliest date on which any material contributing to the batch was removed from the animal, and | ||||
(ii) if the substance was obtained by the growth of organisms on artificial media, the earliest date on which growth was terminated in any of the material contributing to the batch: | ||||
Provided that, in cases where no such test is required or accepted, if a batch of the substance (including all materials contributing to the batch) has for a period of not more than three years been kept in cold storage at a temperature not exceeding 5°C. continuously from the earliest practicable date after that on which the material was removed from the animal or on which growth was terminated in the material, as the case may be, the date of removal from cold storage shall be treated as the date on which the manufacture of the batch is completed. | ||||
|
9 Prohibition of sale of Substance after prescribed date. | 9.—No person shall sell any therapeutic substance after the date recorded on the container, label or wrapper as the date up to which the substance may be expected to retain a potency not less than, or not to acquire a toxicity greater than, that required or permitted by the test, as the case may be: | |||
Provided that a person may at the request of a registered medical practitioner or registered veterinary surgeon sell after the date aforesaid any therapeutic substance (except one that is required to be tested for maximum toxicity) which loses its potency, if he has previously drawn the practitioner's or surgeon's attention to the dates recorded on the container, label or wrapper, and the practitioner or surgeon certifies in writing that the sale is required owing to the urgency of the case. | ||||
|
PART IV.STANDARDS OF STRENGTH, QUALITY AND PURITY AND TESTS FOR DETERMINING WHETHER THOSE STANDARDS HAVE BEEN ATTAINED, UNITS OF STANDARDISATION. | ||||
|
10 Standards. | 10.—Every therapeutic substance intended for sale shall conform with the standards of strength, quality and purity specified in these Regulations, and the tests for determining such conformity shall be applied to samples taken from the final product after every manufacturing process has been completed. | |||
|
11 Tests for Strength and Quality. | 11.—The tests, if any, required for determining the strength and quality of each of the therapeutic substances shall be those set out in the relative Schedule to these Regulations. | |||
|
12 Units of Standardisation. | 12.—The unit of standardisation in relation to a therapeutic substance shall be such unit, if any, as is prescribed by the relative Schedule. | |||
|
13 Tests for Sterility. | 13.—The following tests for the presence of living aerobic or anaerobic bacteria shall be made by the manufacturer or by some institution approved by the Minister for the purpose of carrying out tests on his behalf in the case of— | |||
(a) sera and solutions of serum proteins intended for injection; | ||||
(b) the vaccines to which Part I (A) of the Second Schedule to these Regulations applies; | ||||
(c) toxins, toxoids, and other antigens or mixtures of these or any one of these with serum which are intended to be used in medical or veterinary practice for diagnosis, prophylaxis or treatment by inoculation of the patient or animal; | ||||
(d) solutions of insulin; | ||||
(e) dry preparations of insulin intended for therapeutic use; and | ||||
(f) preparations of the posterior lobe of the pituitary body intended for use by injection, except preparations which, after being sealed in the containers, have been sterilised by heat in a manner satisfactory to the Minister: | ||||
Provided that— | ||||
(i) in the case of dry preparations of insulin the tests shall be applied with such modifications as the Minister considers appropriate; and | ||||
(ii) if a manufacturer satisfies the Minister that he has already in use tests for the presence of living aerobic or anaerobic bacteria, in any of above-named substances, and that these tests, as applied by him, will detect the presence of such bacteria in the substance as ready for issue with a certainty at least equal to that afforded by the application of the tests prescribed by this Part of these Regulations, the Minister may approve the use of such tests in the place of the prescribed tests, but in such a case the Minister may at any time withdraw such approval and require the manufacturer to carry out the prescribed tests. | ||||
|
14 Application of Tests for Sterility. | 14.—The tests shall be applied— | |||
(a) to samples taken from each batch of the substance before the operation of filling and sealing the containers in which it is to be issued has commenced; and | ||||
(b) to the contents of sample containers when ready for issue. | ||||
|
15 Amount of Samples. | 15.—The samples required to be taken under the last preceding Article shall be taken in the following proportions:— | |||
(a) in the case of samples taken from the batch, the quantity taken shall be not less than 0·1 per cent. of the total volume of the batch if the volume is not more than 10 litres, and not less than 10 c.c. if the volume is 10 litres or more, but shall in no case be less than 1 c.c.: | ||||
Provided that if, at the time when the test is made, the batch is contained in a number of bulk containers, samples in the foregoing proportions shall be taken from each of such bulk containers and be separately tested; | ||||
(b) in the case of the contents of sample containers the number of containers taken for test shall be not less than 1 per cent. of the total number filled from the batch if this number is not more than 1,000, and not less than 10 containers if the total number is more than 1,000. | ||||
|
16 Method of preparing and using Media. | 16.—(1) The tests shall be made in fluid media, the quantity of medium contained in each tube or other vessel used in the test being such as to secure that any antiseptic present in the sample is so diluted that the growth of micro-organisms would not be inhibited. | |||
(2) In the case of a test for aerobic organisms the medium shall consist either of a meat extract with the addition of 1 per cent. of peptone, or of such an equivalent as can be prepared by the tryptic digestion of muscle. After the final sterilisation the hydrogen-ion concentration of the medium shall be between the limits represented by pH = 7·2 and pH = 7·8. | ||||
(3) In the case of a test for anaerobic organisms the medium shall consist of a nutrient broth similar to that used in testing for aerobic organisms with the addition of heat coagulated tissue. The tissue shall occupy a depth of not less than 1 centimetre at the bottom of the tube and the broth a depth of not less than 5 centimetres. After the final sterilization the hydrogen-ion concentration of the medium shall be between the limits represented by pH. = 7·2 and pH. = 7·8. In the case of tests of sera, unless the cultures are to be incubated in an apparatus in which the contained atmosphere is devoid of oxygen, the broth shall have above it a layer not less than 1 centimetre in depth of liquid paraffin, vaseline or other suitable material to act as a barrier to the absorption of oxygen. In every case before the test inoculation the medium shall be heated to 100° C for a period sufficient to free it completely from dissolved oxygen and then be cooled to 37° C or lower. | ||||
(4) The Minister may, at the request of any licensee, authorise the use, for the test prescribed under either paragraph (2) or (3) of this Article, of any other specified medium or method of using a specified medium, on being satisfied that its use affords equal certainty in the detection of the presence of living aerobic or anaerobic organisms, as the ease may be. | ||||
|
17 Method of Testing. | 17.—(1) In the case of sample taken from the batch each sample shall be inoculated into tubes or other vessels containing the media, one-half of the total volume of the sample being used for the aerobic and one-half for the anaerobic test. | |||
(2) In the case of the contents of sample containers the contents of each container shall be subjected to the test for aerobic and the test for anaerobic organisms. When the volume in the container is 2 c.c. or more, 1 c.c. shall be used for each test. When the volume in the container is less than 2 c.c., the contents shall be divided into two approximately equal parts, one part being used for the aerobic and the other for the anaerobic test. | ||||
(3) The inoculated tubes shall be incubated at 37° C. for five days and be examined after incubation, permanent records being kept of the examination of each tube. | ||||
|
18 .. | 18.—(1) If at the examination referred to in the last preceding Article no growth of micro-organisms is found in any tube, the sample may be treated as having passed the test. | |||
(2) If at such examination a growth of micro-organisms is visible, further samples may be taken and the tests may be repeated on the further samples so taken; but no container the contents of which form part of the batch shall be issued until such further samples have passed the test. The process of taking samples from the batch for a test may, if necessary, be repeated twice: | ||||
Provided that if the same organism is visible in more than one test, the batch shall be treated as not sterile and the material contained in the batch shall not be issued or used as part of a further batch unless and until it has been re-sterilised and has passed the tests. | ||||
|
19 .. | 19.—Notwithstanding anything contained in the last preceding Article, in any case where— | |||
(a) a substance is certified in writing by a medical practitioner or a registered veterinary surgeon as being required in an emergency, but the licensee has no filled containers in stock; or | ||||
(b) a substance which in the opinion of the Minister is so unstable that the delay occasioned by the completion of the sterility test on filled containers would render its issue in active form impossible, | ||||
the licensee may issue the substance from a batch which has already passed the tests for sterility and freedom from abnormal toxicity, without completing the sterility test on the filled containers, provided that he complies with the following conditions:— | ||||
(i) the licensee shall before the issue take samples in the required proportions from the containers into which the batch is filled, and after the required inoculation and incubation shall examine the tubes every day for five days; | ||||
(ii) if at any examination any growth is visible in any of the tubes, he shall immediately notify the licensing authority; | ||||
(iii) he shall keep available for inspection a record of all issues made under this Article containing such particulars of the circumstances in which the issue is made as the Minister may require. | ||||
|
20 Tests for Freedom from Abnormal Toxicity. | 20.—The following tests for freedom from abnormal toxicity shall, in the case of each batch of serum, be made by the licensee or by some institution approved by the Minister for the purpose of carrying out the tests on his behalf:— | |||
(a) a dose of 0·5 c.c. of the serum shall be injected subcutaneously into a normal mouse and the serum may be treated as having passed the test for freedom from an excess of antiseptic if the injection does not produce death or serious symptoms; and | ||||
(b) a dose of not less than 5 c.c. of the serum shall be injected subcutaneously or intraperitioneally into a normal guinea pig and the serum shall be treated as having passed the test for freedom from other abnormal toxic constituents if the injection does not produce death or serious symptoms. | ||||
|
PART V.LICENCES FOR IMPORT OF THERAPEUTIC SUBSTANCES. | ||||
|
21 .. | 21.—Every applicant for a licence to import a therapeutic substance or substances for purposes other than those of scientific research (in this Part of these Regulations referred to as an "import licence") shall furnish to the Minister a written undertaking with the Minister in a form approved by the Minister, signed by or on behalf of the person who manufactures the therapeutic substance or substances to which the application relates (hereinafter referred to as the "manufacturer"), that he will in the event of such licence being granted during the continuance of the licence comply with the following conditions:— | |||
(a) the applicant shall be the sole accredited agent of the manufacturer for the import of the substance; | ||||
(b) the manufacturer will comply with the conditions imposed on a licensee by paragraphs (a), (b), (d) and (e) inclusive of Article 5 of these Regulations; | ||||
(c) the manufacturer will allow any inspector authorised by the Minister in that behalf to enter, with or without prior notice, any premises where the manufacture of the therapeutic substance is carried on and to inspect the premises and plant and the process of manufacture and the means employed for standardising and testing the substance and to take samples thereof; | ||||
(d) the manufacturer will from time to time report to the Minister any proposal to carry on the manufacture of the therapeutic substance in premises other than those in which it was carried on when the licence was issued and every such change of premises if and when effected and in a case in which the manufacture is carried on in more than one factory any proposal for redistributing functions between factories and every such redistribution if and when effected; | ||||
(e) the manufacturer will comply with the provisions of Part III of these Regulations; | ||||
(f) every substance manufactured by the manufacturer for import under licence will as regards strength, quality, and purity conform with the provisions of Part IV of these Regulations and of the relative Schedule, or, with such standards of strength, quality and purity as may be prescribed for that substance by any Regulations made under the Act and for the time being in operation, or, if no such standards are so prescribed, with such standards of strength, quality and purity as are prescribed in the case of therapeutic substances of a similar class; | ||||
(g) the manufacturer will comply with such further requirements, if any, applicable to manufacturers of therapeutic substances as may be specified in any Regulations which may be made under the Act and of which the Minister has given to the licensee not less than one month's notice. | ||||
|
22 .. | 22.—(1) Every import licence shall be subject to the observance by the manufacturer of the undertaking signed by him in respect of such licence. | |||
(2) Every import licence shall, in addition, be subject to the following conditions:— | ||||
(a) the licensee shall on request furnish to the Minister from every batch of the substance or from such batch or batches as the Minister may from time to time specify, a sample of such amount as the Minister may consider adequate for any examination required to be made; and the licensee shall, if so required, furnish full protocols of the tests which have been applied; | ||||
(b) if the Minister so directs the licensee shall not sell or offer for sale any batch in respect of which a sample is or protocols are furnished under the last preceding sub-paragraph until a certificate authorising the sale of the batch has been issued to him by or on behalf of the Minister; | ||||
(c) the licensee shall, on being informed by the Minister that any part of any batch of the substance has been found by the Minister not to conform with the standards of strength, quality and purity specified in these Regulations and on being directed so to do, withdraw the remainder of that batch from sale and so far as may in the particular circumstances of the case be practicable, recall the issues already made from that batch; | ||||
(d) the licensee shall comply with such further requirements, if any, applicable to the holders of import licences, as may be specified in any Regulations which may be made under the Act and of which the Minister has given to him not less than one month's notice. | ||||
|
PART VI.RESEARCH LICENCES. | ||||
|
23 .. | 23.—Every licence granted to a person engaged in scientific research to import therapeutic substances for the purpose of scientific research (in this Part of these Regulations referred to as a "research licence") shall be subject to the following conditions:— | |||
(a) the licensee shall use the substances imported under the licence for purposes of scientific research exclusively, and shall carry on such research in the place or places specified in the licence or in such other place or places as the Minister may from time to time authorize; | ||||
(b) the licensee shall keep a record of the substances imported under the licence, together with the quantities imported, the date of importation and the name of the manufacturer; | ||||
(c) the licensee shall comply with such further requirements, if any, applicable to the holders of research licences, as may be specified in any Regulations which may be made under the Act, and of which the Minister has given to the licensee not less than one month's notice. | ||||
|
PART VII.THERAPEUTIC SUBSTANCES MANUFACTURED OR IMPORTED FOR EXPORT ONLY. | ||||
|
24 .. | 24.—The Minister may in the case of any particular substance dispense with any of the requirements of these Regulations if he is satisfied that the substance is being manufactured exclusively for sale outside Saorstát Eireann or is being imported exclusively for re-export, and that such dispensation is desirable, regard being had to the nature of any arrangements for regulating the manufacture and sale of the substance in operation in the country to which the substance is to be exported. | |||
Given under the Official Seal of the Minister for Local | ||||
Government and Public Health this 23rd day of | ||||
November, One Thousand Nine Hundred and | ||||
Thirty-Four. | ||||
(Sighnithe) SEÁN T. O CEALLAIGH, | ||||
Minister for Local Government and Public Health. | ||||
|
FIRST SCHEDULE. | ||||
FORM 1. | ||||
Form of Application for a Licence to Manufacture Therapeutic Substances for Sale. | ||||
Therapeutic Substances Act, 1932. | ||||
To Minister for Local Government and Public Health. | ||||
| ||||
hereby apply for a licence to manufacture on premises at the undermentioned therapeutic substances for sale:— | ||||
The names and qualifications of the expert staff responsible for the manufacture or testing of the above-mentioned substances are as follow:— | ||||
| ||||
Signature | ||||
Date | ||||
FORM 2. | ||||
Form of Manufacturer's Licence. | ||||
Therapeutic Substances Act, 1932. | ||||
Licence No. | ||||
The Minister for Local Government and Public Health in exercise of the powers conferred on him by the above-mentioned Act hereby licenses of to manufacture on premises at the undermentioned therapeutic substance(s) for sale:— | ||||
This licence is granted subject to the conditions applicable to manufacturers' licences and to the conditions pertaining to the manufacture of the above-named substance(s) set out in any Regulations made under the said Act and for the time being in force, and may be suspended or revoked by the said Minister under the said Act if the licensee is convicted of an offence under the said Act. | ||||
This licence will, unless previously suspended or revoked by the said Minister, continue in force for a period of two years from the date hereof. | ||||
Given under the Official Seal of the Minister for Local Government and Public Health this day of in the year One thousand nine hundred and | ||||
FORM 3. | ||||
Form of Application for a Licence to Import a Therapeutic Substance. | ||||
Therapeutic Substances Act, 1932. | ||||
To Minister for Local Government and Public Health. | ||||
| ||||
hereby apply for a licence to import the undermentioned Therapeutic substance(s) from | ||||
of being the manufacturer thereof, that is to say:— | ||||
| ||||
Signature | ||||
Date | ||||
FORM 4. | ||||
Form of Import Licence. | ||||
Therapeutic Substances Act, 1932. | ||||
Licence No. | ||||
The Minister for Local Government and Public Health in exercise of the powers conferred on him by the above-mentioned Act hereby licenses of to import the undermentioned therapeutic substance(s) from of , being the manufacturer thereof, that is to say:— | ||||
This licence is granted subject to the conditions pertaining to the importation of the above-mentioned substance(s) set out in any Regulations made under the said Act and for the time being in force and to the observance by such manufacturer of the undertaking signed by him in respect of this licence. | ||||
This licence may be suspended or revoked by the said Minister under the said Act if the licensee is convicted of an offence under the said Act or if there has in the opinion of the said Minister been a breach of the said undertaking. | ||||
This licence will, unless previously suspended or revoked by the said Minister, continue in force for a period of two years from the date hereof. | ||||
Given under the Official Seal of the Minister for Local Government and Public Health this day of One Thousand Nine Hundred and | ||||
FORM 5. | ||||
Form of Application for a research licence to import therapeutic substances for the purpose of scientific research. | ||||
Therapeutic Substances Act, 1932. | ||||
To The Minister for Local Government and Public Health. | ||||
I † of being a person engaged in scientific research, hereby apply for a licence to import the undermentioned therapeutic substances for the purpose of scientific research at the following place or places: | ||||
† The application should be signed by the individual person desiring to import the substances. | ||||
This application is supported by the recommendations appearing below. | ||||
Signature | ||||
Date | ||||
| ||||
1 | ||||
of | ||||
President [Principal, Chairman | ||||
Provost or Vice-Chancellor] of | ||||
2 | ||||
of | ||||
Professor of | ||||
FORM 6. | ||||
Form of Research Licence. | ||||
Therapeutic Substances Act, 1932. | ||||
Licence No. | ||||
The Minister for Local Government and Public Health in exercise of the powers conferred on him by the above mentioned Act, hereby licenses of being a person engaged in scientific research, to import for the purpose of scientific research at | ||||
or in such other place or places as the said Minister may from time to time authorise, the under-mentioned therapeutic substance(s): | ||||
‡The recommendation must be signed by one of the persons named at (a) below and also by a person holding one of the offices named at (b) below:— | ||||
(a) The President of the General Medical Council. | ||||
The President of the Medical Registration Council of Saorstát Eireann. | ||||
The President of the Irish Free State Veterinary Council. | ||||
The Chairman of the Medical Research Council. | ||||
The Provost of Trinity College, Dublin. | ||||
The President of University College, Cork. | ||||
The President of University College, Dublin. | ||||
The President of University College, Galway. | ||||
The Vice-Chancellor, Queen's University, Belfast. | ||||
The President of the Royal College of Physicians, London. | ||||
The President of the Royal College of Physicians of Ireland. | ||||
The President of the Royal College of Physicians, Edinburgh. | ||||
The President of the Royal College of Surgeons of England. | ||||
The President of the Royal College of Surgeons, Edinburgh. | ||||
The President of the Royal College of Surgeons in Ireland. | ||||
The President of the Royal College of Veterinary Surgeons. | ||||
The Principal of the Veterinary College of Ireland, Dublin. | ||||
The President of the Royal Society. | ||||
The President of the Royal Dublin Society. | ||||
The President of the Royal Irish Academy. | ||||
The President of the Royal Academy of Medicine in Ireland. | ||||
The President of the Pharmaceutical Society of Ireland. | ||||
(b) A Professor of Anatomy, Bacteriology, Biochemistry, Hygiene, Materia Medica, Medical Jurisprudence, Medicine, Pathology, Pharmacology, Physiology, Surgery or Therapeutics in a University in the Irish Free State or in a College in the Irish Free State which is incorporated by Charter. | ||||
This licence is granted subject to the conditions pertaining to the importation of therapeutic substances for the purpose of scientific research set out in any Regulations made under the said Act and for the time being in force and may be suspended or revoked by the said Minister if the licensee is convicted of an offence under the said Act. | ||||
This licence will, unless previously suspended or revoked by the said Minister, continue in force for a period of two years from the date hereof. | ||||
Given under the Official Seal of the Minister for Local Government and Public Health this | ||||
day of in the year One thousand nine hundred and | ||||
|
SECOND SCHEDULE. | ||||
VACCINES, TOXINS, TOXOIDS, ANTIGENS, SERA AND ANTITOXINS. | ||||
PART I. | ||||
Vaccines. | ||||
(A.) | ||||
PROVISIONS APPLICABLE TO THE PRODUCTION OF VACCINES. | ||||
Definition. | ||||
1.—(1) This Part of this Schedule applies to vaccines made from any micro-organism pathogenic to man or other animal and to vaccines made from other micro-organisms which have any antigenic value. | ||||
(2) For the purposes of this Part of this Schedule a vaccine means a sterile suspension of a killed culture of the micro-organism from which the vaccine derives its name or a sterile extract or derivative of a micro-organism which has been prepared from a genuine culture of the micro-organism. | ||||
Staff of Establishment. | ||||
2. The establishment where vaccines are prepared must be under the complete direction and control of a competent expert in bacteriology, who must be assisted by a staff adequate for carrying out the tests required during the preparation of the vaccines and in connection with the finished products. | ||||
Proper Name. | ||||
3. The proper name of any vaccine shall be the name of the microorganism from which it is made, followed by the word "vaccine" unless the relative Schedule otherwise provides, or if there is no provision in the relative Schedule some other name is approved by the Minister. Provided that in the case of the under-mentioned preparations the proper name of the vaccine may be as follows:— | ||||
Anti-typhoid vaccine; | ||||
Anti-typhoid-paratyphoid vaccine (T.A.B.); | ||||
Anti-typhoid-paratyphoid-cholera vaccine (T.A.B.C.); | ||||
Anti-plague vaccine; | ||||
Anti-dysentery vaccine; | ||||
Whooping cough vaccine. | ||||
Records. | ||||
4. Cultures used in the preparation of vaccines must, before being manipulated into a vaccine, be thoroughly tested for identity by the generally accepted tests applicable to the particular micro-organism. The permanent records which the licensee is required to keep shall include a record of the origin, properties and characteristics of the cultures. | ||||
Combined Vaccines. | ||||
5. Vaccines may be issued either singly or combined in any proportion in the same container. In the case of combinations of vaccines a name for the combined vaccine may be submitted by the licensee to the Minister, and, if approved, may be used as the proper name of the vaccine. | ||||
Labelling. | ||||
6.—(1) The label on the container shall indicate the composition of the vaccine by reference either:— | ||||
(a) to the number of micro-organisms per c.c.; or | ||||
(b) to the weight of dried substance of micro-organisms per c.c.; or | ||||
(c) to the number of micro-organisms or weight of dried substance of micro-organisms used in preparing 1 c.c. of the finished product, or | ||||
(d) in the case of veterinary products to such methods as are satisfactory to the Minister. | ||||
In the case of a combined vaccine the reference to the number of microorganisms per c.c. or to the weight of dried substance of micro-organism shall distinguish between the several kinds of contributing microorganisms. | ||||
(2) If the vaccine as issued for sale is combined with any substance other than a simple diluent, the exact nature and strength of such substance must be stated on the label. | ||||
Tests. | ||||
7. In the case of any vaccine prepared from a micro-organism which does not grow readily in or on ordinary culture media each batch of the vaccine shall, in addition to being submitted to the general tests for sterility prescribed in Part IV of these Regulations, be tested either in a similar manner in or on media which are specially favourable to the growth of the particular micro-organism from which the vaccine was prepared or by injection into an animal of a species known to be susceptible to infection by the particular organism, and no material from any batch shall be issued unless the batch has passed one of these tests. | ||||
(B.) | ||||
PROVISIONS APPLICABLE TO THE PRODUCTION OF VACCINE LYMPH. | ||||
Definition and Proper Name. | ||||
1. Vaccine Lymph is a preparation of the substance obtained from the vesicles produced on the skin of healthy animals by inoculation of vaccinia virus. Its proper name is "Vaccine Lymph." | ||||
Staff of Establishment. | ||||
2. The establishment in which vaccine lymph is prepared must be under the complete direction and control of a competent expert, who must be assisted by a staff adequate for carrying out the tests required during the preparation of the vaccine lymph and in connection with the finished product. | ||||
Condition and Housing of Animals. | ||||
3.—(1) The animals used in the production of vaccine lymph must be adequately and healthily housed. | ||||
(2) Only healthy animals may be used in the production of vaccine lymph. Each animal intended to be used as a source of vaccine lymph must, before being passed for the production of vaccine lymph, be subjected to a period of observation in quarantine for at least five days. During the period of quarantine the animal must remain free from any sign of disease and must be thoroughly cleaned and groomed. | ||||
Precautions to be Observed in Preparation. | ||||
4.—(1) A special room, with impervious walls and floor, which can be washed and, when necessary, chemically disinfected, must be provided for the inoculation of the animals and the collection of the vaccine lymph. | ||||
(2) The inoculation shall be made on such parts of the animal as are not liable to be soiled by the fæces. The surface used for inoculation shall be shaved and so cleaned as to procure the nearest possible approach to asepsis. | ||||
(3) When the vaccine lymph has been collected, the animal, if not previously killed, shall be killed, and a thorough post-mortem examination of the carcase shall be made by a veterinary surgeon approved by the Minister. A complete record of each such examination shall be kept, and shall be open to inspection by or on behalf of the Minister at any time. If the examination reveals any condition which indicates or suggests that the animal was suffering from any infection (other than vaccinia) the lymph obtained from that animal shall not be issued. | ||||
(4) All instruments and appliances used in the production of vaccine lymph shall be previously subjected to an effective process of sterilisation. | ||||
(5) Laboratories in which vaccine lymph, after removal from the animal, is exposed to the air in the course of the process of preparation must be separated from stables and animal houses by a distance sufficient to avoid the risk of aerial contamination with bacteria from animal excreta. Such laboratories must have impervious walls and floors and must be capable of being readily disinfected when necessary. All processes to which the vaccine lymph is subjected must be such as will preclude the access of bacteria. | ||||
(6) All vaccine lymph must be subjected, after collection, to such treatment with glycerol or other partial disinfectants as will bring its content of bacteria and other microscopically visible micro-organisms within the limit prescribed in paragraph 7 of this Part of this Schedule. | ||||
(7) When the procedures necessary to bring the content of bacteria and other visible micro-organisms within the prescribed limit have been completed, the vaccine lymph shall be kept continuously in cold storage, at a temperature below 0°C., until it is withdrawn to be filled into tubes for issue, after which process the filled tubes shall immediately be returned to cold storage and kept continuously at a temperature below 0°C. until required for issue. | ||||
(8) No vaccine lymph for inoculation into animals shall be imported into Saorstát Eireann unless the Minister is satisfied that it has been tested for the presence of the virus of foot and mouth disease on the mucous membrane of the mouth of a susceptible bovine animal and that the animal remained free from foot and mouth disease for a period of seven days after inoculation. | ||||
Containers. | ||||
5. Vaccine lymph for issue shall be introduced into previously sterilised capillary glass tubes by a method excluding access of bacteria. The tubes shall then be hermetically sealed at each end. Each tube shall contain a quantity of vaccine lymph suitable for the effective vaccination of one human subject: Provided that in case of emergency a larger quantity of vaccine lymph may be issued in containers of larger dimensions, which have been sterilised before the introduction of the lymph, and sealed so as to preclude the access of bacteria. | ||||
Labelling. | ||||
6.—(1) The label on the container or a label or wrapper affixed to the package in which the container is issued for sale shall bear a statement that the potency of the vaccine lymph cannot be assured for more than seven days from the date of completion of manufacture, unless the lymph is kept at a temperature below 10° C. | ||||
(2) For the purpose of Article 8 (3) (c) of these Regulations, the date on which the manufacture of the batch is completed shall be the date on which the vaccine lymph is removed for issue from cold storage after having been kept continuously at a temperature below 0°C., since the date of filling into tubes for issue. | ||||
Tests for purity. | ||||
7.—(1) During the exposure of the vaccine lymph to the action of glycerol or other partial disinfectant under suitable conditions of temperature the number and nature of the living bacteria and other visible micro-organisms present shall be periodically determined, the results being recorded, and the records being kept for inspection. These determinations shall be made by the preparation of plate cultures in the manner provided in sub-paragraph (b) of the next succeeding paragraph and by enumeration of the colonies appearing on incubation for two days at approximately 37° C., and then for at least three days at approximately 20° C. This treatment and examination of the vaccine lymph shall continue until the total number of living bacteria and other visible micro-organisms present has been reduced to not more than 5 in I milligram or 5,000 in 1 c.c. of the vaccine lymph, and no vaccine lymph shall be issued until this reduction has been effected. | ||||
(2) Each batch of vaccine lymph shall be submitted to the following tests for the presence of living gas-producing anaerobic organisms and for the presence of haemolytic streptococci:— | ||||
(a) Test for the presence of living, gas-producing anaerobic organisms.—A sample consisting of not less than 0.1 c.c. of the vaccine lymph shall be inoculated into such a medium and incubated at 37° C. under such conditions as will, to the satisfaction of the Minister, reveal with certainty the presence of living organisms growing and producing gas under anaerobic conditions. If the presence of such organisms is revealed by the test, the batch of lymph containing them shall not be issued. | ||||
(b) Test for the presence of living haemolytic streptococci.—A sample of the vaccine lymph shall be thoroughly mixed with melted nutrient blood agar medium and a culture plate poured from the mixture. This shall be incubated at 37° C., and be examined daily for two days for the presence of colonies having the appearance of colonies of haemolytic streptococci. If any such colony is detected it shall be examined, and the nature of the organism determined. If any such colony is found to be formed of streptococci, the batch of lymph from which it was grown shall not be issued. | ||||
Test for potency. | ||||
8.—(1) Each batch of vaccine lymph, after all the procedures required for preparing it for issue have been completed, shall be tested for potency, so as to ensure its activity at the time of issue. | ||||
(2) For the purpose of a test for potency a dilution shall be prepared by mixing 1 volume of the lymph as diluted for distribution with 1000 volumes of physiological saline solution. The dilution shall be used for the test without filtration. | ||||
(3) The thousandfold dilution of the vaccine lymph shall be tested in accordance with a method approved by the Minister. | ||||
(C.) | ||||
PROVISIONS APPLICABLE TO THE PRODUCTION OF THERAPEUTIC SUBSTANCES CONTAINING LIVING ORGANISMS OR VIRUSES OTHER THAN VACCINE LYMPH. | ||||
1. Every therapeutic substance other than Vaccine Lymph containing, or alleged to contain, living organisms or viruses shall be tested in such manner as the Minister shall approve in each particular case for the purpose of determining— | ||||
(a) that the substance contains in living condition the organism or virus which it is alleged to contain; | ||||
(b) that its administration is reasonably free from danger; | ||||
(c) that it is free from living organisms other than those which it is alleged to contain. | ||||
2. The proper name of such a substance shall be that which the Minister, in each particular case, shall approve in writing. | ||||
PART II. | ||||
Toxins and Antigens. | ||||
(A.) | ||||
PROVISIONS APPLICABLE TO THE REAGENTS USED IN THE SCHICK TEST FOR THE DIAGNOSIS OF SUSCEPTIBILITY TO DIPHTHERIA. | ||||
Definitions and proper names. | ||||
1.—(1) The reagents used in the Schick test are two, Schick Toxin and Schick Control. Their proper names respectively are "Schick Test Toxin" and "Schick Control." | ||||
(2) Schick Test Toxin is a sterile filtrate from a culture on nutrient broth of the specific organism of Diphtheria (Corynebacterium diphtheriae). It may be issued either— | ||||
(a) undiluted, accompanied by a container in the same box or carton holding such a volume of sterile saline solution as, when mixed with the accompanying quantity of the undiluted toxin, will make a dilution of the strength proper for use in the test. The proper name of the substance in this form is "Schick Test Toxin (undiluted)"; or | ||||
(b) already diluted with an appropriate saline solution to the strength proper for use in the test. The proper name of the substance in this form is "Schick Test Toxin (diluted for use)." | ||||
(3) Schick Control is prepared from the same batch of Schick Toxin as that with which it is intended to be used by destroying the specific toxicity. This is effected by heating the toxin in such a manner as to keep it at a temperature not lower than 70° C. for a time not shorter than five minutes. Schick Control is issued in a dilution not weaker than that in which the corresponding toxin is used in the test. | ||||
(4) The dilution of Schick Toxin proper for the test is that in which 0.2 c.c. contains one test dose. | ||||
Tests for potency. | ||||
2. The test dose of Schick Toxin for the purpose of the foregoing provision shall be measured by the following tests:— | ||||
(a) by intracutaneous injection into normal guinea-pigs in mixtures with different proportions of diphtheria antitoxin. One test dose mixed with 1/750th or more of a unit of antitoxin must cause no local reaction, but mixed with 1/1250th or less of a unit of antitoxin must cause a definite local reaction of the type known as the "positive Schick reaction"; | ||||
(b) by intracutaneous injection into normal guinea-pigs, without admixture with antitoxin. 1/50th of 1 test dose must not cause, and ½5th of 1 test dose must cause, a definite local reaction of the type known as the "positive Schick reaction." | ||||
Application of Article 18. | ||||
3. Article 18 of these Regulations shall apply to Schick Toxin (diluted for use) as being a substance so unstable in solution that the delay occasioned by the completion of the sterility test on filled containers prescribed by Part IV of the Regulations would render its issue in active form impossible. | ||||
(B.) | ||||
PROVISIONS APPLICABLE TO DIPHTHERIA PROPHYLACTIC. | ||||
Definition and proper name. | ||||
1. Diphtheria prophylactic is diphtheria toxin (the sterile filtrate from a culture on nutrient broth of Corynebacterium diphtheriæ) or material derived therefrom, the specified toxicity of which has been reduced to a low value by the action of chemical substances. To it may be added diphtheria antitoxin. Its treatment must be such that it retains efficient properties as an immunizing antigen. Its proper name is "Diphtheria Prophlactic." | ||||
No preparation containing unmodified diphtheria toxin shall be issued for the prophlaxis of diphtheria. | ||||
Labelling. | ||||
2. The label on the container shall bear a statement of the dose (hereinafter referred to as the "human dose") appropriate for administration at one injection to a human subject. | ||||
Tests. | ||||
3. Diphtheria Prophylactic shall be submitted to the following tests:— | ||||
(a) Tests to determine that the specific toxicity of the toxin used in its preparation has been so reduced that it does not exceed the prescribed maximum.—Five human doses of the Diphtheria Prophylactic under test shall be injected into each of five normal guinea-pigs each weighing 250 to 350 grammes. This injection must not cause the death of any of the guinea-pigs within six days following the injection. If all the guinea-pigs injected survive for six days but any of them die within thirty days following the injection from the specific toxaemia, one human dose of the Diphtheria Prophylactic under test shall be injected into each of five normal guinea-pigs, each weighing 250 to 350 grammes. This injection must not cause the death of any of the guinea-pigs within 30 days following the injection. | ||||
If a batch of Diphtheria Prophylactic is shown by either of these tests to have a greater toxicity than the maximum hereby indicated, it shall not be issued unless and until the toxicity has been so reduced by further treatment that it does not exceed that maximum. | ||||
(b) Test for potency as an immunizing antigen.—A quantity of Diphtheria Prophylactic not exceeding five human doses shall be injected on one occasion into each of ten normal guinea-pigs; or, alternatively, a quantity of Diphtheria Prophylactic not exceeding one-tenth of a human dose shall be injected into each of ten normal guinea-pigs on each of two occasions, separated by an interval of not more than four weeks. The guinea-pigs shall be tested for immunity to diphtheria toxin, if they have received the single injection hereinbefore prescribed, at a date not later than six weeks after injection, and if they have received the two injections hereinbefore prescribed, at a date not later than three weeks after the second injection. | ||||
The test for immunity may be made by either of the two following methods:— | ||||
(i) by intracutaneous injection into each guinea-pig of one test dose of Schick Toxin. If more than two out of the ten guinea-pigs exhibit a positive Schick reaction, the batch of Diphtheria Prophylactic shall be treated as insufficiently potent, and shall not be issued; or | ||||
(ii) by subcutaneous injection into each guinea-pig of five minimum lethal doses of diphtheria toxin. If more than two out of the ten guinea-pigs die as the result of this injection the batch of Diphtheria Prophylactic shall be treated as insufficiently potent, and shall not be issued. | ||||
(C.) | ||||
PROVISIONS APPLICABLE TO TUBERCULINS AND OTHER PREPARATIONS FROM THE BACILLUS TUBERCULOSIS AND ITS CULTURES INTENDED FOR HUMAN USE. | ||||
(Note.—The name "tuberculin" has been frequently applied to any extract, suspension or other preparation of the Bacillus tuberculosis or of media on which that bacillus has been cultivated. In the following Part of this Schedule the name is used in a more restricted sense and applies only to tuberculins as therein defined.) | ||||
Tuberculins. | ||||
Definition and proper name. | ||||
1.—(1) Tuberculins are preparations of fluid media on which the Bacillus tuberculosis has been grown in artificial culture and which have been freed by filtration from the bacilli. | ||||
(2) For the purposes of this Schedule tuberculins are classified in two groups (a) Old Tuberculin, and (b) Tuberculin Bouillon Filtrate. | ||||
Old Tuberculin. | ||||
2.—(1) Old Tuberculin is the concentrated filtrate from the growth of Bacillus tuberculosis on a suitable nutrient broth. For its preparation the bacillus must be grown at approximately 37° C. for a period, usually not less than six weeks, sufficient to allow the surface of the fluid medium to become covered by a thick growth of the bacillus. At the end of this period the fluid medium, from which the bacilli may or may not have been previously separated by filtration, must be concentrated by evaporation to one-tenth of its original volume, and then be filtered. If the required test for potency shows that the preparation so concentrated is more potent than the standard preparation, the potency may be reduced to the required degree by appropriate dilution. If the test shows that the potency is less than that of the standard preparation, it shall not be increased by further evaporation. The proper name of the preparation is "Old Tuberculin," with or without a suffix such as T., or P.T. The suffix T., if used, will indicate that the bacillus used in preparing the Tuberculin was of the human type, and the suffix P.T. that the bacillus used was of the bovine type. | ||||
(2) The standard preparation of Old Tuberculin is a quantity of Old Tuberculin kept in an institution approved by the Minister. | ||||
(3) Each batch of Old Tuberculin shall be tested for potency by observation of its specific toxicity, by a method approved by the Minister, in such a way that the potency of the preparation under test is measured by comparison with that of the standard preparation. Old Tuberculin shall not be issued if its activity differs from that of the standard preparation to such an extent that the difference is revealed by the test. | ||||
(4) Each batch of Old Tuberculin shall be tested for the absence of non-specific toxicity by the subcutaneous, injection of 0·5 c.c. into a normal guinea-pig, and shall be treated as having passed the test if such injection does not cause death or serious symptoms. | ||||
Tuberculin Bouillon Filtrate. | ||||
3.—(1) Tuberculin Bouillon Filtrate is the unconcentrated Filtrate from the growth of Bacillus tuberculosis on a suitable nutrient broth. For its preparation the bacillus must be grown at approximately 37° C. for a period usually not less than six weeks, sufficient to allow the surface of the fluid medium to become covered by a thick growth of the bacillus. At the end of this period the medium is freed from bacilli by filtration through a bacteria-proof filter. The proper name of the preparation is "Tuberculin Bouillon Filtrate," with or without a suffix such as T.O.A. or P.T.O. The suffix T.O.A., if used, will indicate that the bacillus used in preparing the Tuberculin Bouillon Filtrate was of the human type, and the suffix P.T.O. will indicate that the bacillus used was of the bovine type. | ||||
(2) Each batch of Tuberculin Bouillon Filtrate shall be tested for the absence of non-specific toxicity by the subcutaneous injection of 5 c.c. into a normal guinea-pig, and shall be treated as having passed the test if such injection does not cause death or serious symptoms. | ||||
Test for sterility. | ||||
4. All tuberculins shall be tested for sterility in accordance with Articles 13 to 17 of Part IV of these Regulations. Tuberculin Bouillon Filtrate shall be tested in addition for absence of living tubercle bacilli by a method satisfactory to the Minister. | ||||
Tubercle Vaccines. | ||||
Definition and proper name. | ||||
5. Tubercle vaccines are preparations made from the bacillary substance obtained by growth of the Bacillus tuberculosis on artificial media, and consisting of suspensions of the killed organisms, or of products therefrom, in water or other suitable suspending fluids. The proper name is "Tubercle Vaccine," and any other descriptive title or symbol indicating the origin of the bacilli or the nature of the process of preparation must be used in addition to, and not in substitution for, the name "Tubercle Vaccine." | ||||
Application of provisions as to bacterial vaccines. | ||||
6. The provisions of Part I (A) of this Schedule (which relates to the production of bacterial vaccines) shall apply to the production of tubercle vaccines. | ||||
(D.) | ||||
PROVISIONS APPLICABLE TO TUBERCULINS INTENDED FOR VETERINARY USE. | ||||
1. Tuberculins for veterinary use are classified in three groups:— | ||||
(a) Tuberculin prepared from mammalian type of Bacillus tuberculosis grown on a fluid nutrient medium and subsequently prepared in the manner laid down for "Old Tuberculin" in the second Schedule, Part II (C) 2 of these Regulations. | ||||
(b) Tuberculin bouillon filtrate as defined in the second Schedule, Part II (C) 3 of these Regulations. | ||||
(c) Tuberculin prepared from the avian type of the Bacillus tuberculosis grown on a nutrient fluid medium and subsequently prepared in the manner laid down for "Old Tuberculin" in the second Schedule, Part II (C) 2 of these Regulations. The proper name of the preparation is Tuberculin with the suffix A. | ||||
Test for Sterility. | ||||
2. Each batch of tuberculin for veterinary use in the foregoing classification shall be tested for sterility in accordance with Articles 13—17 Part 4 of these Regulations and in addition for the absence of living tubercle bacilli by a method satisfactory to the Minister. | ||||
(E.) | ||||
PROVISIONS APPLICABLE TO MALLEIN. | ||||
1. Mallein is the concentrated or unconcentrated filtrate from a growth of Bacillus mallei in a fluid nutrient medium. For this preparation the bacillus must be grown at approximately 37° C. for a period of not less than three weeks. | ||||
At the end of this period the medium is heated to 100° C. for one hour and freed from bacilli by filtration through a bacteria proof filter. | ||||
The proper name of the preparation is "Mallein" with or without the suffix C. The suffix C. if used will indicate that the filtrate has been concentrated by evaporation to one-tenth of its original volume. | ||||
Test for Sterility. | ||||
Each batch of mallein shall be tested for sterility in accordance with Articles 13—17 of Part 4 of these Regulations, and in addition for the absence of living glanders bacilli by a method satisfactory to the Minister. | ||||
PART III. | ||||
PROVISIONS APPLICABLE TO THE PRODUCTION OF ALL SERA FROM LIVING ANIMALS. | ||||
Condition and housing of animals. | ||||
1.—(1) The animals used in the production of sera must be adequately and healthily housed. | ||||
(2) Only healthy animals shall be used in the preparation of sera, and in particular the presence of glanders in horses or other equidae and of tuberculosis in cattle must be excluded by testing with mallein and tuberculin respectively. | ||||
(3) Every new animal intended to be used as a source of serum must be subjected to a period of observation in quarantine for at least seven days, before being admitted to the stables in which the serum-yielding animals are housed. | ||||
(4) Every animal used as a source of serum must either be actively immunized against tetanus toxin or must be passively immunized against that toxin by injections of tetanus antitoxin in such doses as to ensure the constant presence of that antitoxin in the blood during the whole period of the use of the animal as a source of serum. | ||||
Staff of Establishment. | ||||
2. The establishment must be under the complete direction and control of a competent expert in bacteriology and serology, assisted by a staff adequate for carrying out the tests required during the preparation of the sera and in connection with the finished products. | ||||
Precautions to be observed in preparation. | ||||
3.—(1) Laboratories where sera are exposed to the air in the course of the process of preparation must be separated by a sufficient distance from stables and animal houses to avoid the risk of aërial contamination with bacteria from animal excreta, and must be rendered fly-proof to prevent such contaminations by insects. Such laboratories must have impervious walls and floors and must be capable of being readily disinfected when necessary. | ||||
(2) A special room with impervious walls and floor which can be washed and, when necessary, chemically disinfected must be provided for the collection of blood from the living animal. | ||||
(3) An efficient system of manure removal must be used, which will prevent its accumulation in the vicinity of any room where blood or serum is collected or handled. | ||||
(4) An adequate number of efficient sterilizers must be provided for the sterilization of all glass-ware or other apparatus with which the serum may come into contact in the course of its preparation. | ||||
(5) All processes to which the serum is subjected during and after its collection from the animal, must be designed to preserve its sterility, but in the case of artificially concentrated sera, it shall suffice that the process of concentration is conducted with scrupulous cleanliness and in such a manner as to avoid unnecessary or dangerous contamination. | ||||
(6) The laboratories in which the testing of the sera for potency, sterility and freedom from abnormal toxicity are carried out must be adequate for the purpose. An adequate supply of animals for use in such tests and suitable housing for such animals must be provided. | ||||
(7) Provision must be made for complying with any special conditions which may be laid down in these Regulations relating to the production and issue of the particular serum, in respect of which the licence is granted. | ||||
Unhealthy or infected animals. | ||||
4. In an animal used in the production of serum is found to be suffering from an infection, except one produced by living organism against which it is being immunized, or shows signs of serious or persistent ill-health not reasonably attributable to the process of immunization, the licensee shall immediately report the matter to the Minister and shall, if the Minister orders an inspection and the inspector so directs, cause such animal to be killed and a post mortem examination of it to be made by a veterinary surgeon, and take steps to prevent any serum obtained from the animal being sold or offered for sale until permission is given by the Minister. If the result of the post mortem examination is such as to bring under suspicion the health of any of the other animals used for the production of serum, the Minister may prohibit the use of those animals for the production of serum or may take such other steps as may be necessary to prevent the issue of serum which may be dangerous to human or animal health: | ||||
Provided that in a case of emergency the person in charge of the establishment may order the destruction of an animal used in the production of serum and suspected of infection, and shall in that case give notice forthwith to the Minister and shall permit an inspector to be present at the post mortem examination. | ||||
PART IV. | ||||
Provisions applicable to particular Sera and Antitoxins. | ||||
(A.) | ||||
PROVISIONS APPLICABLE TO ANTI-BACTERIAL SERA, ANTITOXIC SERA AND OTHER ANTI-SERA FOR WHICH NO POTENCY TEST IS PRESCRIBED. | ||||
(Note.—The sera and antitoxins to which this Part of this Schedule applies are the sera or solutions of the purified proteins of sera separated from the blood of animals which have been artificially immunized against cultures of one or more organisms or against a soluble toxin or toxins produced by the organism or organisms or against antigenic substances prepared from the organism or organisms.) | ||||
Proper Name. | ||||
1. The proper name of any serum prepared against a micro-organism to which Division A of this Part of this Schedule applies shall be the recognised scientific name of the organism or some generally recognised abbreviation thereof, preceded by the prefix "anti," and followed by the word "serum," as, for example, "anti-meningococcus serum." The proper name of any antitoxic serum may be formed from the word "antitoxin," preceded by the name of the organism from which the toxin was prepared, and followed, if desired, by a term indicating the source or the strain of that organism, for example, "streptococcus antitoxin (Scarlatina)." | ||||
Quality. | ||||
2.—(1) Any such serum shall be issued for therapeutic use in the form of either— | ||||
(a) natural serum, i.e., the fluid obtained from coagulated blood or plasma of the immunised animal without any addition, other than antiseptic, or subtraction; or | ||||
(b) a solution of the purified serum proteins containing the specific antibodies. | ||||
(2) At the time of issue, the liquid shall be clear or show at most a slight opalescence or precipitate. Preparations of the natural serum or plasma shall not contain more than 10 per cent. of solid matter. A solution of the serum protein shall not contain more than 20 per cent. of solid matter. | ||||
Labelling. | ||||
3.—(1) The label on the container shall indicate the total number of c.cs. in the container. | ||||
(2) The label on the container or the label or wrapper on the package shall indicate the nature of the particular product, that is to say, whether natural serum or a solution of the purified serum proteins. | ||||
Cultures. | ||||
4. The cultures used in immunizing the animals shall be at all times open to inspection, and specimens shall be furnished for examination at the request of the Minister. | ||||
Records. | ||||
5.—(1) The permanent records which the licensee is required to keep shall include the following particulars— | ||||
(a) as to the cultures— | ||||
(i) the source from which the culture was obtained; | ||||
(ii) the nature of the material from which the culture was isolated, and the date of its isolation; and | ||||
(iii) evidence of the identity and specificity of the culture; | ||||
(b) as to the procedure used in immunizing the animals— | ||||
(i) the method of preparing the culture or antigen used for immunization; | ||||
(ii) the dosage and methods employed in administering the culture or antigen; | ||||
(iii) the period in the course of immunization at which blood is withdrawn for preparation of the serum; | ||||
(c) any tests which may have been applied to the serum to determine its content of specific antibodies or its specific therapeutic potency. | ||||
(2) If the licensee desires to treat the performance of any test recorded under sub-paragraph (1) (c) of this paragraph as determining the date of completion of manufacture for the purposes of Article 8 of these Regulations, he shall submit full particulars of the proposed test to the Minister and obtain the approval of the Minister. | ||||
(B.) | ||||
PROVISIONS APPLICABLE TO ANTI-DYSENTERY SERUM (SHIGA) AND OTHER ANTI-DYSENTERY SERA. | ||||
Anti-dysentery Serum (Shiga). | ||||
Proper Name. | ||||
1. Anti-dysentery serum (Shiga) is the serum, or the globulins containing the specific immune substances, separated from the blood of animals which have been immunized against the toxins, cultures or bacterial substances obtained by artificial culture of the Bacillus dysenteriae (Shiga). The proper name of the substance is "Anti-dysentery Serum (Shiga)." | ||||
Standard preparation. | ||||
2. The standard preparation is a quantity of dried serum, obtained from horses immunized against the toxic constituents of the Bacillus dysenteriæ (Shiga), and kept in an institution approved by the Minister. | ||||
Quality. | ||||
3.—(1) Anti-dysentery serum (Shiga) shall be issued for therapeutic use in the form of either— | ||||
(a) the natural serum, i.e., the fluid obtained from the coagulated blood or plasma of the immunized animals without any addition other than antiseptic, or subtraction; or | ||||
(b) the solution of the globulins containing the specific immune substances; or | ||||
(c) a dry powder prepared from (i) the natural serum or (ii) the globulins containing the specific immune substances. | ||||
(2) If issued in fluid form the liquid shall, at the time of issue, be clear or show, at most, a very slight opalescence or precipitate. Preparations of the natural serum or plasma shall not contain more than 10 per cent. of total solid matter. A solution of the separated antitoxic globulins shall not contain more than 20 per cent. of total solid matter. | ||||
Strength. | ||||
4.—(1) The potency of anti-dysentery serum, with respect to its content of antibodies for the toxic constituents of the Bacillus dysenteriae (Shiga) shall be determined by intravenous injection into mice of mixtures of the serum with a solution or suspension of the said toxic constituents, which solution or suspension has been standardised in relation to the standard preparation of anti-dysentery serum. | ||||
(2) Each container of anti-dysentery serum (Shiga) shall contain a sufficient number of units in excess of the minimum total number of units indicated on the label to ensure that the said minimum total number of units will still be present in the container at the date appearing on the label pursuant to Article 8 (3) (e) of these Regulations as the date up to which the preparation may be expected to retain its potency. | ||||
Unit of Standardization | ||||
5. The unit of anti-dysentery serum (Shiga) for the purposes of these Regulations is the specific neutralising activity for the toxic constituents of the Bacillus dysenteriæ (Shiga) contained in such an amount of the standard preparations as the Minister may from time to time indicate as the quantity exactly equivalent to the unit accepted for international use. | ||||
Labelling. | ||||
6.—(1) The label on the container shall indicate— | ||||
(a) the minimum total number of units in the container; and | ||||
(b) either (i) the potency of the preparation with respect to its antitoxic value for the toxic constituents of the Bacillus dysenteriae (Shiga), expressed as the minimum number of units per c.c. in the case of liquid products, or as the minimum number of units per gramme in the case of dry products; or | ||||
(ii) the total number of c.c. in the container. | ||||
(2) The label on the container or the label or wrapper on the package shall indicate the nature of the particular product, that is to say, whether natural serum, or a solution of the globulins containing the specific immune substances, or a dried natural serum or dried globulins. | ||||
Other Anti-dysentery Sera. | ||||
Proper names. | ||||
7. Anti-dysentery sera prepared by immunizing animals against bacilli producing dysentery in man, other than the B. dysentariae (Shiga), shall conform with the provisions of Division (A) of this Part of this Schedule which are applicable to sera for which no potency test is prescribed. The proper name shall in each case be "Anti-dysentery Serum," followed, in brackets, by the name or symbol by which the particular strain or strains of dysentery bacilli are identified by bacteriologists—as, for example, "Anti-dysentery Serum (Flexner)," "Anti-dysentery Serum (Y)," "Anti-dysentery Serum (Flexner, Y)." | ||||
8. A mixed anti-dysentery serum, prepared by immunizing animals against the B. dysenteriae (Shiga) and in addition against one or more of the other bacilli associated with human dysentery, shall conform with the provisions of Division (A) of this Part of this Schedule, and shall also, with respect to its content of immune substances for the B. dysenteriae (Shiga) and its products, conform with paragraphs 3, 4, 5 and 6 (2) in Division (B) thereof; and the number of units shown on the label shall indicate the neutralizing value of the serum for the products of the B. dysenteriae (Shiga) only. The proper name of such a serum shall be "Anti-dysentery Serum," followed, in brackets, by the names or symbols indicating the strains used in its preparation, as, for example—"Anti-dysentery Serum (Shiga, Flexner, Y)." | ||||
(C.) | ||||
PROVISIONS APPLICABLE TO DIPHTHERIA ANTITOXIN. | ||||
Definition and Proper Names. | ||||
1. Diphtheria antitoxin is the serum or the antitoxic globulins separated from the blood of animals which have been immunized against diphtheria toxin. When the serum or antitoxic globulins are obtained from the blood of horses or other equidae, the proper name of the substance is "diphtheria antitoxin." When the serum or antitoxic globulins are obtained from animals other than horses or other equidae, the proper name is "diphtheria antitoxin" followed by the common name of the animal from which the substance is prepared. | ||||
Standard preparation. | ||||
2. The standard preparation is a quantity of dried diphtheria antitoxin kept in an institution approved by the Minister. | ||||
Strength. | ||||
3.—(1) Diphtheria antitoxin having a potency of less than 400 units per c.c. in the case of liquid preparations, or less than 4,000 units per gramme in the case of dried preparations, shall not be issued. | ||||
(2) Each container of diphtheria anti-toxin shall contain a sufficient number of units in excess of the minimum total number of units indicated on the label to ensure that the said minimum total number of units will still be present in the container at the date appearing on the label pursuant to Article 8 (3) (e) of these Regulations as the date up to which the preparation may be expected to retain its potency. | ||||
Quality. | ||||
4.—(1) Diphtheria antitoxin shall be issued for therapeutic and prophylactic use in the form of either— | ||||
(a) the serum, i.e., the fluid obtained from the coagulated blood or plasma of animals immunized against diphtheria toxin without any addition other than antiseptic, or subtraction; or | ||||
(b) the solution of the globulins containing the specific antitoxin; or | ||||
(c) a dry powder prepared from (i) the natural serum or (ii) the antitoxic globulins and containing no antiseptic or other added substance. | ||||
(2) If issued in fluid form the liquid at the time of issue shall be clear or shall show, at most, a very slight opalescence or precipitate. Preparations of the natural serum or plasma shall not contain more than 10 per cent. of solid matter. A solution of the separated antitoxic globulins shall not contain more than 0·1 gramme of solid matter for each 500 units. | ||||
Unit of standardisation. | ||||
5. The unit of diphtheria antitoxin for the purposes of these Regulations is the specific neutralizing activity for diphtheria toxin contained in such an amount of the standard preparation as the Minister may from time to time indicate as the quantity exactly equivalent to the unit accepted for international use. | ||||
Test for potency. | ||||
6. The potency in units of diphtheria antitoxin shall be determined in accordance with a method approved by the Minister by the injection into guinea-pigs of a mixture consisting of the antitoxin under test and of a diphtheria toxin which has been standardized in relation to the standard preparation. | ||||
Labelling. | ||||
7.—(1) The label on the container shall indicate— | ||||
(a) the minimum total number of units in the container; and | ||||
(b) either (i) the potency of the preparation expressed as the minimum number of units of antitoxin per c.c. in the case of liquid products, or as the minimum number of units of antitoxin per gramme in the case of dry products; or | ||||
(ii) the total number of c.cs. in the container. | ||||
(2) The label on the container or the label or wrapper on the package shall indicate the nature of the particular product, that is to say, whether natural serum, a solution of antitoxic globulins, dried natural serum, or dried antitoxic globulins. | ||||
(D.) | ||||
PROVISIONS APPLICABLE TO TETANUS ANTITOXIN. | ||||
Proper name. | ||||
1. Tetanus antitoxin is the serum, or the antitoxic globulins, separated from the blood of animals which have been immunized against tetanus toxin. The proper name of the substance is "Tetanus antitoxin." | ||||
Standard preparation. | ||||
2. The standard preparation is a quantity of dried tetanus antitoxin kept in an institution approved by the Minister. | ||||
Strength. | ||||
3.—(1) Tetanus antitoxin having a potency of less than 300 units per c.c. in the case of liquid preparations, or less than 3,000 units per gramme in the case of dried preparations, shall not be issued for prophylaxis in man or for prophylaxis or treatment in animals. | ||||
Tetanus antitoxin having a potency of less than 1,600 units per c.c. in the case of liquid preparations, or less than 16,000 units per gramme in the case of dried preparations, shall not be issued for the treatment of tetanus in man. | ||||
(2) Each container of tetanus antitoxin shall contain a sufficient number of units in excess of the minimum total number of units indicated on the label to ensure that the said minimum total number of units will still be present in the container at the date appearing on the label pursuant to Article 8 (3) (e) of these Regulations as the date up to which the preparation may be expected to retain its potency. | ||||
Quality. | ||||
4.—(1) Tetanus antitoxin shall be issued for therapeutic and prophylactic use in the form of either— | ||||
(a) the serum, i.e., the fluid obtained from the coagulated blood or plasma of animals immunized against the tetanus toxin without any addition other than antiseptic, or subtraction; or | ||||
(b) the solution of the globulins containing the specific antitoxin; or | ||||
(c) a dry powder prepared from (i) the natural serum or (ii) the antitoxic globulins, and containing no antiseptic or other added substance. | ||||
(2) If issued in fluid form the liquid at the time of issue shall be clear or show at most a very slight opalescence or precipitate. Preparations of the natural serum or plasma shall not contain more than 10 per cent. of total solid matter. A solution of the separated antitoxic globulins shall not contain more than 0·1 gramme of solid matter for each 600 antitoxin units. | ||||
Unit of standardisation. | ||||
5. The unit of tetanus antitoxin for the purposes of these Regulations is the specific neutralizing activity for tetanus toxin contained in such an amount of the standard preparation as the Minister may from time to time indicate as the quantity exactly equivalent to the unit accepted for international use*. | ||||
*This unit is one-half of the unit established in the United States of America under the authority of an Act of the 1st July, 1902. | ||||
| ||||
Test for potency. | ||||
6. The potency in units of tetanus antitoxin shall be determined by the subcutaneous injection into guinea-pigs or mice of mixtures of the preparation with a tetanus toxin which has been standardised in relation to the standard preparations of tetanus antitoxin. The neutralizing value may be determined by observation either— | ||||
(a) of the greatest dose which fails to protect a guinea-pig or mouse from death within 4 days, or | ||||
(b) of the least dose which suffices to protect a mouse or guinea-pig from the appearance of symptoms of tetanus. | ||||
Labelling. | ||||
7.—(1) The label on the container shall indicate— | ||||
(a) the minimum total number of units in the container; and | ||||
(b) either (i) the potency of the preparation expressed as the minimum number of units of antitoxin per c.c. in the case of liquid products, or as the minimum number of units of antitoxin per gramme in the case of dry products; or (ii) the total number of c.cs. in the container; and | ||||
(c) A statement† that the numbers of units indicated are equivalent to one-half of those numbers of American units. | ||||
(2) The label on the container or the label or wrapper on the package shall indicate the nature of the particular product, that is to say, whether natural serum, a solution of antitoxic globulins, dried natural serum, or dried antitoxic globulins. | ||||
(E.) | ||||
PROVISIONS APPLICABLE TO GAS-GANGRENE ANTITOXIN (PERFRINGENS). | ||||
Proper name. | ||||
1. Gas-Gangrene Antitoxin (perfringens) is the serum, or the antitoxic globulins, separated from the blood of animals which have been immunised against the specific toxin prepared by the growth of Bacillus perfringens (B. welchii) in a fluid medium. The proper name of the substance is "Gas-Gangrene Antitoxin (perfringens)". | ||||
Standard Preparation. | ||||
2. The standard preparation is a quantity of dried gas-gangrene antitoxin (perfringens) kept in an institution approved by the Minister. | ||||
Quality. | ||||
3.—(1) Gas-gangrene antitoxin shall be issued for therapeutic use in the form of either— | ||||
(a) the serum, i.e., the fluid obtained from the coagulated blood or plasma of the immunised animals without any addition other than antiseptic, or subtraction; or | ||||
(b) the solution of the globulins containing the specific immune substances; or | ||||
(c) a dry powder prepared from (i) the natural serum or (ii) the globulins containing the specific immune substances. | ||||
† The statement may be conveniently given in arithmetical form, thus, for example:—"2,000 units (= 1,000 American units)." | ||||
(2) If issued in fluid form the liquid shall, at the time of issue, be clear or show, at most, a very slight opalescence or precipitate. Preparations of the natural serum or plasma shall not contain more than 10 per cent. of solid matter. A solution of the separated antitoxic globulins shall not contain more than 20 per cent. of total solid matter. | ||||
Strength. | ||||
4.—(1) The potency in units of gas-gangrene antitoxin (perfringens) shall be determined, in accordance with a method approved by the Minister, by the injection into animals of a mixture of the antitoxin under test with a gas-gangrene (perfringens) toxin which has been standardised in relation to the standard preparation of gas-gangrene antitoxin (perfringens). | ||||
(2) Each container of gas-gangrene antitoxin (perfringens) shall contain a sufficient number of units in excess of the minimum total number of units indicated on the label to ensure that the said minimum total number of units will still be present in the container at the date appearing on the label pursuant to Article 8 (3) (e) of these Regulations as the date up to which the preparation may be expected to retain its potency. | ||||
Unit of Standardisation. | ||||
5. The unit of gas-gangrene antitoxin (perfringens) for the purposes of these Regulations is the specific neutralizing activity for gas-gangrene (perfringens) toxin contained in such an amount of the standard preparation as the Minister may from time to time indicate as the quantity exactly equivalent to the unit accepted for international use. | ||||
Labelling. | ||||
6.—(1) The label on the container shall indicate— | ||||
(a) the minimum total number of units in the container; and | ||||
(b) either (i) the potency of the preparation expressed as the minimum number of units of antitoxin per c.c. in the case of liquid products or as the minimum number of units of antitoxin per gramme in the case of dry products; or | ||||
(ii) the total number of c.cs. in the container. | ||||
(2) The label on the container or the label or wrapper on the package shall indicate the nature of the particular product, that is to say, whether natural serum, a solution of antitoxic globulins, dried natural serum or dried antitoxic globulins. | ||||
Mixed Antitoxins. | ||||
7. A mixed antitoxin, containing antitoxins against other toxins than that of the Bacillus perfringens, shall, with respect to its content in units of gas-gangrene antitoxin (perfringens), conform with paragraphs 4, 5 and 6 of this Part of this Schedule. | ||||
|
THIRD SCHEDULE. | ||||
ARSPHENAMINE (COMMONLY KNOWN AS SALVARSAN) AND ITS DERIVATIVES. | ||||
PART I. | ||||
GENERAL PROVISIONS APPLICABLE TO ARSPHENAMINE AND TO ITS DERIVATIVES. | ||||
Standard preparation. | ||||
1. The standard preparations of arsphenamine and of the derivatives thereof are quantities of those preparations kept in an institution approved by the Minister. | ||||
Biological tests. | ||||
2.—(1) The tests shall be carried out either— | ||||
(a) in an institution approved by the Minister; or | ||||
(b) if the Minister so directs in the laboratories of the licensee. | ||||
(2) The licensee shall, if the Minister so directs, transmit to the approved institution for testing a sample from each finished batch of arsphenamine, or its derivative, intended for issue. The sample shall consist of at least six sealed containers of the product as completed for issue, taken by random sampling from the whole batch, and each containing at least 0.6 gramme of the product. If the Minister directs that the tests shall be carried out in the laboratories of the licensee, they shall be carried out in strict accordance with the directions given by the Minister, and in comparison with the standard preparation of arsphenamine or the derivative thereof corresponding to the product under test. | ||||
(3) The tests shall consist of the following:— | ||||
(a) Test for maximum toxicity.—Several separate containers from each finished batch shall be tested for toxicity by intravenous (or, where the Part of this Schedule relating to a particular derivative requires, by subcutaneous) injection into at least ten mice and five rats, or into such number of animals of some other species as the Minister may consider equivalent, and no batch shall be passed for issue which shows a toxicity greater than that of the standard preparation when tested under identical conditions. The test shall be conducted in accordance with such detailed instructions as the Minister may issue. | ||||
(b) Test for therapeutic potency.—Samples from each batch shall be tested for therapeutic potency on a series of mice or rats infected with a suitable strain of pathogenic trypanosomes (T. brucei, T. equiperdum, &c.) in accordance with the following general method and with such detailed instructions as the Minister may issue:— | ||||
(i) the mice or rats on which the test is made shall be infected with the trypanosome employed to an equal degree, the degree being determined by enumeration per unit volume of blood; | ||||
(ii) samples from each batch shall be tested by means of several doses each of which shall be administered to at least three of the animals, and the result shall be evaluated by comparison with the effects of the standard preparation, administered to animals of the same species, having the same degree of infection. | ||||
Method of issue. | ||||
3. Arsphenamine and any derivative of arsphenamine shall be issued in the form of a dry powder, the glass container being filled before being sealed with some inert gas to the exclusion of oxygen, unless permission is given by the Minister for the issue of a particular derivative in some other form. | ||||
PART II. | ||||
SPECIAL PROVISIONS APPLICABLE TO ARSPHENAMINE. | ||||
Proper name. | ||||
1. Arsphenamine is the dihydrochloride of dioxy-diamino-arseno-benzene. Its proper name is "Arsphenamine." | ||||
Quality. | ||||
2. Arsphenamine must have the following physical and chemical characteristics:— | ||||
(a) the substance must be in the condition of a pale yellow to yellow, dry, amorphous powder, freely mobile in contact with glass surfaces, and without odour, except that due to traces of ether; | ||||
(b) if 0.5 gramme of the powder is added to 35 c.c. of distilled water, it must dissolve completely within 15 minutes, yielding a clear pale yellow solution, not acid in reaction to Congo-red paper, and perfectly clear and free from flocculi, hairs, dust and suspended particles of every kind. When 1 c.c. of a 15 per cent. aqueous solution of sodium hydroxide is added to this solution of arsphenamine, it should cause a preliminary separation of the insoluble arseno base, which, if the mixture is gently agitated, should redissolve in the excess of alkali to form a clear, bright yellow solution. This solution, when diluted to 250 c.c. with a 0.5 per cent. solution of pure sodium chloride in distilled water, should give a clear light yellow solution; | ||||
(c) the dry powder, as taken directly from the sealed ampoules in which it is issued, must contain not less than 30 per cent., or more than 34 per cent. of arsenic, as determined by a method approved by the Minister. | ||||
Test for stability. | ||||
3. The product, as filled into ampoules, shall be kept at a temperature of 56° C. for at least 24 hours, and shall retain its colour, physical properties and solubility, as specified in paragraph 2, substantially unchanged at the end of that period. | ||||
PART III. | ||||
SPECIAL PROVISIONS APPLICABLE TO NEOARSPHENAMINE. | ||||
Proper name. | ||||
1. Neoarsphenamine is the sodium salt of dioxy-diamino-arseno-benzene-methylene-sulphoxylic acid. Its proper name is "Neoarsphenamine." | ||||
Quality. | ||||
2. Neoarsphenamine must have the following physical and chemical characteristics:— | ||||
(a) the substance must be in the condition of a yellow, dry powder, freely mobile in contact with glass surfaces, and without odour, except such as is due to traces of ether or alcohol; | ||||
(b) the substance must be soluble in water, but insoluble in absolute ethyl alcohol and in ether. If 0.6 gramme of the substance is added to 1 cubic centimetre of distilled water, it must dissolve rapidly and completely and form a clear, yellow solution, mobile and free from gelatinous particles and suspended matter of every kind; | ||||
(c) a normal solution of sodium carbonate or a 5 per cent. solution of the anhydrous carbonate, added in equal volume to a 10 per cent. aqueous solution of neoarsphenamine, must not produce a precipitate; | ||||
(d) diluted hydrochloric acid (B.P.) added in equal volume to a 10 per cent. aqueous solution of neoarsphenamine must give a yellow precipitate of the free acid from neoarsphenamine. If the mixture is warmed, sulphur dioxide must be evolved so as to be detected by iodate-starch paper; | ||||
(e) when a solution of 0.2 gramme of neoarsphenamine in 10 c.c. of water is acidified with phosphoric acid and distilled to about one-half its volume, formaldehyde must be evolved so as to be detected in the distillate by a red ring formed at the line of contact when five drops of a 1 per cent. solution of phenol is added and a layer of sulphuric acid is run under the mixture; | ||||
(f) the dry powder, as taken directly from the ampoules in which it is issued, must contain not less than 18 per cent. nor more than 21 per cent. of arsenic, as determined by a method approved by the Minister. | ||||
Test for stability. | ||||
3. The product as filled into ampoules shall be kept at a temperature of 56° C. for at least 24 hours and shall retain its colour, physical properties and solubility substantially unchanged at the end of that period. | ||||
PART IV. | ||||
SPECIAL PROVISIONS APPLICABLE TO SULPHARSPHENAMINE. | ||||
Proper name. | ||||
Quality. | ||||
2. Sulpharsphenamine must have the following physical and chemical characteristics:— | ||||
(a) the substance must be in the condition of a yellow, dry powder, freely mobile in contact with glass surfaces, and without odour, except that due to traces of ether or alcohol; | ||||
(b) the substance must be soluble in water but insoluble in alcohol and in ether. If 0.6 gramme of the substance is added to 1 c.c. of distilled water, it must dissolve rapidly and completely, and form a clear, yellow solution, mobile and free from gelantinous particles and suspended matter of every kind; | ||||
(c) a normal solution of sodium carbonate or a 5 per cent. solution of the anhydrous carbonate, added in equal volume to a 10 per cent. aqueous solution of sulpharsphenamine must not produce a precipitate; | ||||
(d) five volumes of diluted hydrochloric acid (B.P.) added to one volume of a 10 per cent. aqueous solution of sulpharsphenamine must give, after a few minutes, a yellow precipitate of the free acid from sulpharsphenamine. If the mixture is boiled, sulphur dioxide must be evolved so as to be detected by iodate-starch paper; | ||||
(e) when a solution of 0.2 gramme of sulpharsphenamine in 10 c.c. of water is acidified with phosphoric acid and distilled to about one-half its volume, formaldehyde must be evolved so as to be detected in the distillate by a red ring formed at the line of contact when five drops of a 1 per cent. solution of phenol is added and a layer of sulphuric acid is run under the mixture; | ||||
(f) on addition of an equal volume of 1 in 10,000 indigo-carmine solution, a 10 per cent. watery solution of sulpharsphenamine must not reduce the indigo-carmine in 5 minutes at 50° C; | ||||
(g) the dry powder, as taken directly from the ampoules in which it is issued, must contain not less than 18 per cent. or more than 21 per cent. of arsenic, as determined by a method approved by the Minister; | ||||
Tests for toxicity and therapeutic potency. | ||||
3. The test for maximum toxicity and for therapeutic potency prescribed in paragraph 2 (3) of Part 1 of this Schedule shall, in the case of sulpharsphenamine, be carried out by subcutaneous injection into mice or rats. | ||||
Test for stability. | ||||
4. The product as filled into ampoules shall be kept at 56ø C. for at least 24 hours and shall retain its colour, physical properties and solubility substantially unchanged at the end of that period. | ||||
PART V. | ||||
SPECIAL PROVISIONS APPLICABLE TO DERIVATIVES OF ARSPHENAMINE OTHER THAN THOSE SPECIFIED IN PARTS II AND III OF THIS SCHEDULE. | ||||
Nature of substance. | ||||
1. In the case of any derivative of arsphenamine other than those specified in Parts II, III and IV of this Schedule the applicant for a manufacturer's or an import licence shall submit to the Minister with his application a statement of the true chemical nature and composition of the derivative, and a full and detailed account of the chemical tests by which that composition is determined and by which the uniformity of successive batches is secured. | ||||
Proper name. | ||||
2. The applicant shall also submit with his application the name which he proposes to use for the derivative to which the application relates, and such name, if approved by the Minister, may be used as the proper name of the derivative. | ||||
Chemical tests. | ||||
3. If a licence is granted for the manufacture of such a derivative of arsphenamine, the licensee shall carry out on each batch of the derivative such, if any, of the chemical tests submitted with the application as are accepted by the Minister, and any others which the Minister may direct as requisite for determining the composition and securing its uniformity. No batch of the derivative which fails to pass any of the tests so accepted or directed shall be issued. | ||||
Tests for toxicity and potency. | ||||
4. Each batch of such derivative shall further be tested, by biological methods, for toxicity and potency, according to the methods prescribed in Part I of this Schedule. In the event of no standard preparation being available for a particular derivative, the tests shall be made in such form and their results interpreted in accordance with such criteria as the Minister may direct. | ||||
|
FOURTH SCHEDULE. | ||||
INSULIN. | ||||
Proper name. | ||||
1. Insulin is the preparation of the specific antidiabetic principle of the pancreas. Its proper name is "Insulin." | ||||
Special conditions of licence. | ||||
2. It shall be a condition of every licence to manufacture or to import insulin:— | ||||
(a) that it shall not be issued in a mixture with any other therapeutic agent except with the previous consent of the Minister; | ||||
(b) that if issued for injection suspended in some medium in which it is not itself soluble, it shall be tested before suspension. | ||||
Standard preparation. | ||||
3. The standard preparation is a quantity of dry soluble insulin hydrochloride prepared and kept in an institution approved by the Minister. | ||||
Unit of standardisation. | ||||
4. The unit of insulin for the purposes of these Regulations is the specific activity contained in such an amount of the standard preparation as the Minister may from time to time indicate as the quantity exactly equivalent to the unit accepted for international use. | ||||
Quality. | ||||
5. The acidity of the prepared watery solution, as determined by a suitable indicator, shall be such that the hydrogen-ion concentration is not less than that corresponding to pH=4, or greater than that corresponding to pH=3. | ||||
Tests. | ||||
6.—(1) The methods used for testing the potency of preparations in comparison with the standard preparation shall be such as the Minister may from time to time approve. | ||||
(2) In addition, samples from each batch shall be tested in such manner as the Minister may direct for the purpose of ascertaining its stability under ordinary conditions of storage. | ||||
Container. | ||||
7. In the case of a prepared solution of insulin the glass of the container shall be non-alkaline resistance glass. | ||||
Labelling. | ||||
8. In the case of a prepared solution of insulin the label on the container shall indicate the strength as the number of units per c.c., and in the case of compressed tablets as the number of units in each tablet. | ||||
|
FIFTH SCHEDULE. | ||||
_________ | ||||
PITUITARY (POSTERIOR LOBE) EXTRACT. | ||||
Proper Name. | ||||
1. Pituitary extract is the watery extract prepared from the separate posterior lobe of the pituitary body, or the watery solution of one or more of the separated active principles of that lobe. The proper name of the complete watery extract is "Pituitary (posterior lobe) Extract." The proper name of a solution containing one of the separated active principles is "Oxytocic principle of the pituitary posterior lobe" or "Pressor principle of the pituitary posterior lobe" or such other name descriptive of such a solution as the Minister may in any particular case approve in writing. | ||||
Standard preparation. | ||||
2. The standard preparation is a quantity of dried acetone-extracted substance obtained from the posterior lobes of fresh pituitary bodies of oxen. This standard is kept in an institution approved by the Minister. | ||||
Unit of standardisation. | ||||
3.—(1) The unit of pituitary extracts for the purposes of these Regulations is the specific activity corresponding to that yielded by 0.5 milligramme of the standard preparation when extracted by the method approved by the Minister under this Schedule. | ||||
(2) When the preparation is a solution of a separated active principle, the unit employed in indicating the strength shall be the amount of that active principle yielded to extraction by 0.5 mgm. of the standard preparation as determined by the appropriate biological test. | ||||
Quality. | ||||
4. The acidity of the prepared watery extract shall be such that the hydrogen-ion concentration is not less than that corresponding to pH = 4, or greater than that corresponding to pH = 3. | ||||
Tests. | ||||
5.—(1) The method used for preparing the extract from the standard preparation and for its use in a comparative biological test and the biological methods employed in making the test shall be such as the Minister may from time to time approve. | ||||
(2) Samples from each batch of the finished product shall be tested for sterility in accordance with the methods set forth in Part IV of these Regulations, unless the finished product has been sterilized by heat in a manner satisfactory to the Minister after being sealed in the containers. | ||||
Container. | ||||
6. The glass of the container shall be non-alkaline resistance glass. | ||||
Labelling. | ||||
7. The label on the container shall indicate the strength of the extract as the number of units per c.c. | ||||
8. The date to be specified in compliance with the requirements of Article (8) (3) (e) of these Regulations shall be such date as the Minister shall in any particular case have approved in writing. |